Genetics of Myotonic Dystrophy & FSHD
Myotonic dystrophy and FSHD result from an abnormal expansion of a certain part of DNA (deoxyribonucleic acid). DNA is made up of chemicals that provide “instructions” to make proteins and to carry out the functions of all life. These instructions or genes are contained in certain sections of DNA. Genes determine such things as hair color, eye color or other more complex traits. Humans have about 30,000 genes, with countless combination to shape who we are!
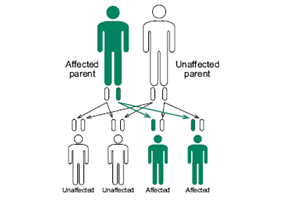 These instructions or genes are passed on from generation to generation. We receive half of our genes from our mother and half from our father. If one parent has the affected DM or FSHD gene, each child has 50% chance of inheriting the condition. The gender of the child does not change the likelihood of inheriting the condition. If a child does not receive the abnormal gene, they will not develop or pass on the disease.
These instructions or genes are passed on from generation to generation. We receive half of our genes from our mother and half from our father. If one parent has the affected DM or FSHD gene, each child has 50% chance of inheriting the condition. The gender of the child does not change the likelihood of inheriting the condition. If a child does not receive the abnormal gene, they will not develop or pass on the disease.
Genetic tests are available for myotonic dystrophy (types 1 and 2) and FSHD. Additionally, prenatal and preimplantation diagnosis are available to determine before birth or conception if a baby has inherited the condition. More specific information about DM1, DM2, and FSHD are below.
Genetic Testing for FSHD
Facioscapulohumeral muscular dystrophy (FSHD) is a genetic condition that results from a DNA mutation. The mutation is a DNA deletion or a decrease in the amount of DNA that is normally present on a chromosome. There is a genetic test available for FSHD, although it is still unknown how the mutation results in FSHD or which genes are affected.
Over 95% of individuals with FSHD have a form that researchers now refer to as FSHD type 1 (FSHD1). This form of FSHD results from the loss (deletion) of a specific segment of DNA at the tip of chromosome 4. DNA and proteins are tightly packaged together in chromosomes in cells of the body. Humans have 23 pairs of chromosomes that help regulate and enable our bodies to have over 30,000 genes!
The genetic test for FSHD locates and measures the size of DNA deletion on chromosome 4. DNA is extracted from the blood sample and a chemical is used to break the DNA into smaller sections. The sections are placed into a gel with an electrical current that causes the pieces of DNA to move through the gel based upon their size. The size of the sections of DNA are compared to pieces of a known size. The test has a 95% rate of accuracy in detecting a genetic mutation.
| Condition |
Size of DNA Deletion
(in kilobase pairs) |
| Normal |
Greater than 51 base pairs |
| Borderline |
Between 36 and 50 base pairs |
| Affected |
Less than 35 base pairs |
There are three possible outcomes for this test. The presence of the DNA deletion indicates that the person will likely develop some symptoms of FSHD during their life, although approximately 5% of individuals with the deletion do not develop symptoms. Research has shown an association between the size of the DNA deletion and the severity of FSHD symptoms.
If the deletion is not detected, it indicates that the individual has not inherited the condition. They will not develop symptoms of FSHD nor will they pass the gene onto their children.
Over the past several years, scientists have found that in about 5% of individuals with FSHD, there is no deletion or loss of DNA on chromosome 4. For these individuals, a DNA test will show a negative result, suggesting that they do not have FSHD. When the sample is further analyzed with specialized research techniques, scientists will, instead, find a change in the chemical structure of the DNA in the same region of chromosome 4 that results in an unfurling of the DNA structure. This unraveling of DNA causes a second form of FSHD that is now referred to as “FSHD2.” These patients have a loosened DNA structure on chromosome 4 which activates the DUX4 gene, like in FSHD type 1. Researchers have recently discovered the gene (SMCHD1) that causes FSHD Type 2.
Both FSHD1 and FSHD2 appear to have identical clinical manifestations and scientists believe the disease-causing mechanism is the same in both, despite the different changes on chromosome 4. Tests for FSHD2 are only available in research settings at the present time. If you have questions about your DNA results and whether further testing would be beneficial for you, please talk to your physician or genetic counselor.
Genetic Testing for Myotonic Dystrophy Type 1
Myotonic Dystrophy is a genetic condition that results from a DNA mutation. The mutation is a DNA expansion or an increase in the amount of DNA that is normally located on a chromosome. The additional DNA is located on chromosome 19. The mutation affects the gene for dystrophia myotonica protein kinase (DMPK) that results in abnormal clumps inside individual cells, which prevents cells in muscles and other tissues from functioning normally. Within the DMPK gene there is a section of DNA that contains a repeated sequence of three DNA nucleotide bases, CTG. It is not uncommon to have a small number of CTG repeats within the DMPK gene; too many repeats will result in a disruption of cellular functioning.
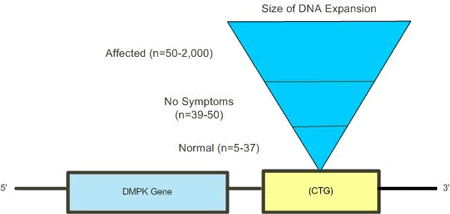
The genetic test for DM locates the specific area on chromosome 19 and measures the size of the DNA expansion. This test has nearly a 100% rate of accuracy in detecting the genetic mutation.
| Condition |
Size of DNA Deletion
(in CTG repeats) |
| Normal |
Less than 38 repeats |
| Borderline |
Between 39 and 49 repeats |
| Affected |
Mild 50 to approximately 150
Classic approximately 100 to 1000
Congenital greater than 2000 |
There are three possible outcomes for this DNA test. A positive result confirms the presence of the inherited DNA expansion. Positive test results indicate that a person will develop some symptoms of DM during their life. There is a relationship between the size of the expansion and the age of onset and severity of Myotonic dystrophy.
A negative test result indicates that a person has not inherited the DNA expansion. They will not develop symptoms of DM nor will they pass the gene onto their children. There is also the possibility of receiving inconclusive results if the size of the DNA expansion is between the normal and abnormal ranges. In general, the person will not develop DM but still has the chance of passing the mutation to their children.
Genetic Testing for Myotonic Dystrophy Type 2
Myotonic Dystrophy type 2 (DM2) is a genetic condition that results from a mutation in your DNA. The mutation referred to as a DNA expansion is an increase in the amount of DNA that is normally located on a chromosome. The additional DNA is located on chromosome 3. The mutation affects the gene for zinc finger protein 9 (ZnF9) which prevents cells in muscles and other tissues from functioning normally. Within the ZnF9 gene there is a section of DNA that contains a repeated sequence of four DNA nucleotide bases, CCTG. It is normal to have a small number of CCTG repeats within gene. If there are too many repeats it will affect various cell functions.
The genetic test for DM2 is designed to isolate and measure the size of the DNA expansion on chromosome 3. This test has nearly a 100% rate of accuracy in detecting the genetic mutation.
| Condition |
Size of DNA Deletion
(in kilobase pairs) |
| Normal |
Less than 175 base pairs |
| Borderline |
Between 177-372 base pairs |
| Affected |
Greater than 372 base pairs |
The number of CCTG base pairs in affected individuals averages approximately 20,000. There are three possible outcomes for this DNA test. A positive result confirms the presence of the inherited DNA expansion. Positive test results indicate that a person will develop symptoms of DM2I during their life. A negative test results indicates that a person has not inherited the DNA expansion. They will not develop symptoms of DM nor will they pass the gene on to their children. There is also the possibility of receiving inconclusive results if the size of the DNA expansion is between the normal and abnormal ranges. In general, the person will not develop DM but may still have the chance of passing the mutation to their children.