Edwards (Trisomy 18)
Clinical History
A 39-year-old woman is in her 17th week of gestation. A screening ultrasound reveals fetal anomalies. A therapeutic abortion is done and tissue is sent for cytogenetics.
Gross Examination
The gross examination of the fetus shows a foot length of 2.4 cm, which is consistent with 17 weeks gestation. The feet bilaterally show rocker bottom morphology. At the caudal end of the spinal column a defect is noted with a thin membrane overlying the cord.
Microscopic Examination
A lumbrosacral myelomeningocele is confirmed. The vertebral body is incomplete at the point of the defect. The squamous epithelium is not continuous overlying the vertebral column; instead there is immature neural tissue in continuity with the squamous epithelium.
Click an image to view larger format.
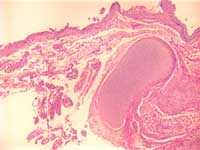
Figure 1: Lumbrosacral myelomenigocele
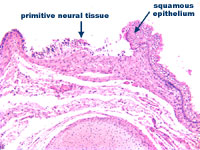
Figure 2: Lumbrosacral myelomenigocele
at higher power
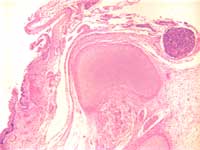
Figure 3: Incomplete formation of
vertebral body with primitive neural
tissue in continuity with squamous
epithelium.
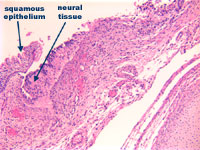
Figure 4: Incomplete formation of
vertebral body with primitive neural
tissue in continuity with squamous
epithelium at higher power
Cytogenics
Karyotype analysis showed a female fetus with 47, XX, +18/46, XX karyotype. This is consistent with an Edward syndrome (trisomy 18) mosaicism. The possibility of maternal contamination causing the two karyotypes cannot be ruled out.
Discussion
The incidence of trisomy 18 is 1/3,500 to 1/7000 live births, with an increased incidence with advancing maternal age. It is the second most common trisomy syndrome after Down's (trisomy 21). Females account for 80% of the cases. It usually results from nondisjunction occurring in maternal meiosis II. A full trisomy is involved in 95% of the cases. Genotypic mosaicism may result from a postzygotic mitotic nondisjunction. The phenotype will be variable depending on the number of cells affected. A translocation can also produce the syndrome. The phenotype can again be variable here, however if the region 18q11-q12 is present in triplet then the most serious trisomy 18 anomalies will be present.
Most (95%) of trisomy 18 cases die in embryonic or fetal life. Of those that are born less that 10% will survive beyond 1 year of age. Most will die of a congenital anomaly.
A variety of clinical presentations can be seen. The most commonly seen entities include failure to thrive, developmental retardation, hypertonia, overlapping fingers, rocker bottom feet, short sternum, congenital heart disease, and cryptorchidism. The case presented here has the characteristic rocker bottom feet. In addition there is a lumbrosacral myelomeningocele. This is one of the many central nervous system malformations that can be seen, others include: microcephaly, cerebellar hypoplasia, meningoencephalocele, anencephaly, hydrocephaly, holoprosencephaly, Arnold Chiari malformation, hypoplasia or aplasia of the corpus callosm, defective falx cerebri, frontal lobe defects, migration defects and arachnoid cysts.
Dr. Enzo Fallone, June 2002