Pediatrics / Steiner Lab / Current Research Projects / Analyses of the Beta-Thalassemia Modifier Genes BCL11a and HSB1L-MYB
Genomic and Functional Analyses of the Beta-Thalassemia Modifier Genes BCL11a and HSB1L-MYB
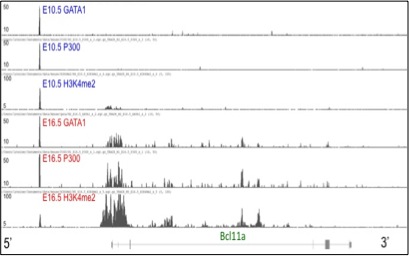
Chromatin Immunoprecipitation coupled with high throughput sequencing (ChIP-seq) demonstrates significant differences
in factor binding and chromatin structure at the BCL11a locus in primitive and definitive erythroid precursors.
The goal of this project is to identify critical regulatory elements of BCL11a and HSB1L-MYB, two genetic regions important for determining disease severity in beta-thalassemia.
In beta-thalassemia, patients who have higher levels of fetal hemoglobin have less severe disease.1 BCL11a and MYB are expressed at specific time points in red blood cell development and suppress the expression of fetal hemoglobin. Multiple studies have demonstrated that people with sequence variants in the BCL11a or HSB1L-MYB genes have higher levels of fetal hemoglobin2 and less severe beta-thalassemia.3 The majority of these sequence variants occur outside of the protein coding regions of these genes, and likely disrupt critical genetic regulatory elements.
Identification and characterization of the critical regulatory elements of these genes will enhance our prediction of disease severity and provide novel targets for therapeutic intervention.
References:
-
Thein, S.L. Genetic modifiers of the beta-haemoglobinopathies. Br J Haematol 141, 357-66 (2008).
-
Menzel, S. et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 39, 1197-9 (2007).
-
Pandit, R.A. et al. Association of SNP in exon 1 of HBS1L with hemoglobin F level in beta0-thalassemia/hemoglobin E. Int J Hematol 88, 357-61 (2008).