Investigating the Role of Extracellular Matrix Alteration(s) in Macular Degeneration
Collaborators: Dr. Bela Anand-Apte, Dr. John Crabb, Dr. Alex Hewitt, Dr. Alice Pebay, Dr. David Williams
Age-related macular degeneration and related macular dystrophies lead to loss of central vision and are a major cause of blindness. In a subset of these maculopathies including AMD, the retinal pigment epithelium-extracellular matrix (RPE-ECM) is the primary site of disease pathology. Several studies have implicated defective ECM turnover due to impaired activity of matrix metalloproteinases (MMPs) and tissue-inhibitors of MMPs (TIMPs) in the development of drusen and choroidal neovascularization (CNV) in these maculopathies. Particularly, altered expression and activity of the ECM-secreted TIMP3 protein and its target MMPs (MMP2, MMP9, MMP14) have been correlated with development of macular degeneration. However, the precise effects of TIMP3 and/or MMP dysregulation on RPE cell function and their consequences in maculopathies are not known. Importantly, dissecting the effect of TIMP3 or MMP dysfunction on a specific cell type, RPE, and its role in disease development has been complicated by the fact that RPE-choroid complex acts as a functional unit in vivo.
Human induced pluripotent stem cells (hiPSCs) provide a unique platform to interrogate pathology and disease-associated physiology in an individual cell type using patient’s own cells. This is particularly relevant to RPE, as multiple studies have shown that hiPSC-derived RPE cells display key physical and functional attributes of human RPE cells in vivo. In fact, we have demonstrated that hiPSC-RPE in culture lay a basement membrane, secrete important ECM and ECM regulating proteins and utilized the relatively long culture life of hiPSC-RPE to age the cells and assist the development of drusen and ECM protein accumulation (COL4) in patient-derived hiPSC-RPE cells from patients with Sorsby’s fundus dystrophy (SFD), a macular dystrophy caused by mutations in TIMP3 gene (Galloway et al PNAS 2017, see figure below).
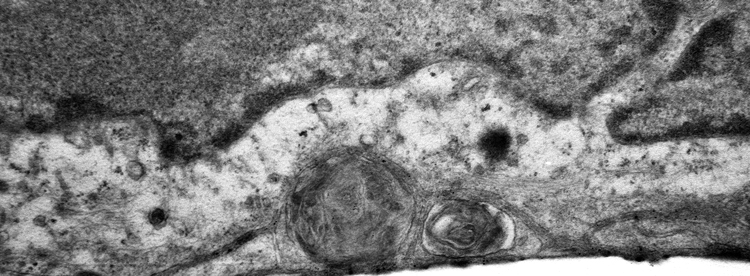
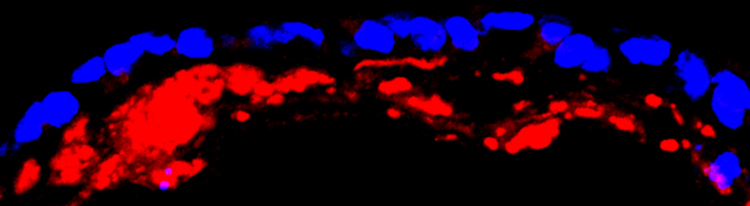
Electron microscopy (top) and confocal microscopy (below) analyses of patient-derived SFD hiPSC-RPE demonstrate presence of sub-RPE drusen-like deposits (the central hallmark of SFD and similar maculopathies) that contain major constituents of drusen in the human eye including APOE (shown in red in the confocal image). Galloway et al PNAS 2017
In this project, we are utilizing hiPSC-RPE derived from SFD patients (that display key phenotypic feature of the disease in a dish, see above and Galloway et al PNAS 2017 ) and unaffected controls to delineate the molecular and subsequently pathological consequences of TIMP3 dysfunction in macular degeneration.