Research Projects
Investigating the Role of Extracellular Matrix Alteration(s) in Macular Degeneration
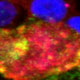 Age-related macular degeneration and related macular dystrophies lead to loss of central vision and are a major cause of blindness. In a subset of these maculopathies including AMD, the retinal pigment epithelium-extracellular matrix (RPE-ECM) is the primary site of disease pathology. Several studies have implicated defective ECM turnover due to impaired activity of matrix metalloproteinases (MMPs) and tissue-inhibitors of MMPs (TIMPs) in the development of drusen and choroidal neovascularization (CNV) in these maculopathies.
Age-related macular degeneration and related macular dystrophies lead to loss of central vision and are a major cause of blindness. In a subset of these maculopathies including AMD, the retinal pigment epithelium-extracellular matrix (RPE-ECM) is the primary site of disease pathology. Several studies have implicated defective ECM turnover due to impaired activity of matrix metalloproteinases (MMPs) and tissue-inhibitors of MMPs (TIMPs) in the development of drusen and choroidal neovascularization (CNV) in these maculopathies.
Determining the Role of CLN3 Gene/Protein in the Human Retina
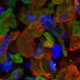 Mutations in CLN3 gene cause Juvenile Neuronal Ceroid Lipofuscinosis (JNCL; Battens disease; CLN3), a lysosomal storage disorder that lead to progressive neurological dysfunction and retinal degeneration. In fact, vision loss associated with retinal degeneration is one of the first and prominent clinical features of JNCL. Despite this, very little about the function of CLN3 gene in the retina is known. Previous studies have shown that CLN3 is an integral membrane protein that undergoes N-linked glycosylation and phosphorylation at multiple sites. However, the function and subcellular localization of CLN3 gene/protein in mammalian cells is debated. Animal models of CLN3, CLN3-/- mice exist and display increased autofluorescence accumulation in the retina, decreased ERG b-wave amplitudes, reduced cone photoreceptor function and loss of INL cell density and ganglion cell death.
Mutations in CLN3 gene cause Juvenile Neuronal Ceroid Lipofuscinosis (JNCL; Battens disease; CLN3), a lysosomal storage disorder that lead to progressive neurological dysfunction and retinal degeneration. In fact, vision loss associated with retinal degeneration is one of the first and prominent clinical features of JNCL. Despite this, very little about the function of CLN3 gene in the retina is known. Previous studies have shown that CLN3 is an integral membrane protein that undergoes N-linked glycosylation and phosphorylation at multiple sites. However, the function and subcellular localization of CLN3 gene/protein in mammalian cells is debated. Animal models of CLN3, CLN3-/- mice exist and display increased autofluorescence accumulation in the retina, decreased ERG b-wave amplitudes, reduced cone photoreceptor function and loss of INL cell density and ganglion cell death.
Learn more about Determining the Role of CLN3 Gene/Protein in the Human Retina
Delineating the Shared Molecular Mechanism of Maculopathies
 Macular degenerative diseases are complex disorders that lead to loss of central vision. In a subset of these maculopathies, including age-related macular degeneration (AMD), disruption of the RPE-ECM complex has been implicated to cause the disease pathology. Furthermore, histopathological and clinical studies have demonstrated that some inherited maculopathies share extensive phenotypic similarities with each other as well as with AMD, suggesting that a similar disease mechanism may underlie these varied dystrophies.
Macular degenerative diseases are complex disorders that lead to loss of central vision. In a subset of these maculopathies, including age-related macular degeneration (AMD), disruption of the RPE-ECM complex has been implicated to cause the disease pathology. Furthermore, histopathological and clinical studies have demonstrated that some inherited maculopathies share extensive phenotypic similarities with each other as well as with AMD, suggesting that a similar disease mechanism may underlie these varied dystrophies.
Learn more about Delineating the Shared Molecular Mechanism of Maculopathies
Elucidating the molecular basis of vision loss in Battens disease
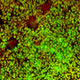 Human induced pluripotent stem cell (hiPSC) technology and genome editing capabilities are scientific advances that have already, independently, revolutionized our approach to studying and treating human eye diseases. However, it is the integration of these technologies and the ability to study the disease pathophysiology and test potential drug-treatments in patient-derived cells in vitro in parallel with a patient’s own eyes in vivo which promotes the biggest advances in the field of translational vision research and therapeutics. For example, a prominent limitation of histopathologic studies in human eyes is that the postmortem retina is obtained at a late stage of the disease, making it difficult to differentiate between initial insult and secondary effects.
Human induced pluripotent stem cell (hiPSC) technology and genome editing capabilities are scientific advances that have already, independently, revolutionized our approach to studying and treating human eye diseases. However, it is the integration of these technologies and the ability to study the disease pathophysiology and test potential drug-treatments in patient-derived cells in vitro in parallel with a patient’s own eyes in vivo which promotes the biggest advances in the field of translational vision research and therapeutics. For example, a prominent limitation of histopathologic studies in human eyes is that the postmortem retina is obtained at a late stage of the disease, making it difficult to differentiate between initial insult and secondary effects.
Learn more about Elucidating the molecular basis of vision loss in Battens disease