Delineating the Shared Molecular Mechanism of Maculopathies
Collaborators: Dr. Bela Anand-Apte, Dr. John Crabb, Dr. Alex Hewitt, Dr. Alice Pebay, Dr. David Williams
Macular degenerative diseases are complex disorders that lead to loss of central vision. In a subset of these maculopathies, including age-related macular degeneration (AMD), disruption of the RPE-ECM complex has been implicated to cause the disease pathology. Furthermore, histopathological and clinical studies have demonstrated that some inherited maculopathies share extensive phenotypic similarities with each other as well as with AMD, suggesting that a similar disease mechanism may underlie these varied dystrophies. For instance, Sorsby’s fundus dystrophy (SFD), and Doyne honeycomb retinal dystrophy (DHRD), two inherited macular dystrophies, similar to AMD, can lead to 1) bilateral loss of central vision due to choroidal neovascularization (CNV) and/or RPE atrophy, 2) prolonged dark adaptation and 3) reduced EOG response. Furthermore, histopathologic studies in donor eyes of SFD, DHRD and AMD patients have shown abnormal lipid-rich sub-RPE deposits (drusen) that stain strongly for TIMP3 and/or TIMP3-EFEMP1 complexes. However, because the postmortem retina from macular degeneration patients is obtained at a late stage of the diseases, differentiating between initial insult and secondary effects is difficult. Thus far, macular degeneration research has mainly employed either primary or immortalized RPE cells, heterologous expression systems or genetic mouse models and we have gained significant insights about the biological process(s) affected in macular degeneration from these studies. hiPSC-derived retinal cells provide another suitable platform to recapitulate the sequence of molecular and pathologic events in patient-derived cells. In fact, we have demonstrated that multiple molecular and pathological alterations consistent with the AMD phenotype, including complement pathway alteration, extracellular matrix protein accumulation, drusen formation, neovascularization can be mimicked in a hiPSC-derived cell model of macular degeneration ( unpublished data in the lab; (Galloway et al PNAS 2017, also see figure below).

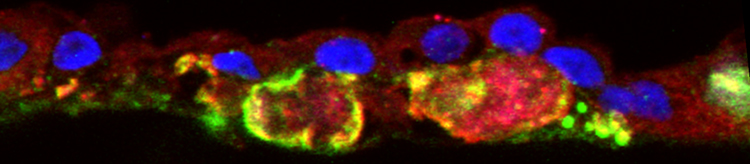
Electron micoscopy (top) and Confocal microscopy (below) analyses of patient-derived hiPSC-RPE demonstrate presence of sub-RPE drusen-like deposits (the central hallmark of SFD and similar maculopathies) that contain major constituents of drusen (APOE: red, TIMP3, green) in the human eye. Galloway et al PNAS 2017
In this project, we are utilizing hiPSC-derived human cell models of AMD and related macular dystrophies (already established in the laboratory) to establish the consequence of specific molecular alteration on phenotypic manifestations of the disease in patient’s own cells in a time course study and thereby identify “common mechanistic defects” that are central to the development of AMD-like pathology.