Determining the Role of CLN3 Gene/Protein in the Human Retina
Collaborators: Dr. Jonathan Mink, Dr. David Pearce, Dr. Jill Weimer, Dr. David Williams
Mutations in CLN3 gene cause Juvenile Neuronal Ceroid Lipofuscinosis (JNCL; Battens disease; CLN3), a lysosomal storage disorder that lead to progressive neurological dysfunction and retinal degeneration. In fact, vision loss associated with retinal degeneration is one of the first and prominent clinical features of JNCL. Despite this, very little about the function of CLN3 gene in the retina is known. Previous studies have shown that CLN3 is an integral membrane protein that undergoes N-linked glycosylation and phosphorylation at multiple sites. However, the function and subcellular localization of CLN3 gene/protein in mammalian cells is debated. Animal models of CLN3, CLN3-/- mice exist and display increased autofluorescence accumulation in the retina, decreased ERG b-wave amplitudes, reduced cone photoreceptor function and loss of INL cell density and ganglion cell death. The widespread retinal pathology affecting multiple retinal cell type(s) is consistent with the human disease. However, the disease in the CLN3-/- mice is delayed compared to humans with prominent phenotypic and functional alterations appearing in 12-24 month old animals. This is a huge disparity given the average life span of mice ranges between 2-3 years. Furthermore, CLN3 knockout model may not be representative of the disease because recent studies have suggested that truncated CLN3 protein retains some of its functional properties. Given the disparity of the CLN3 mouse models with the human disease, it is likely that CLN3 does not play the same role in the rodent vs. human retina. Therefore, studying the role of CLN3 in human derived retinal cell model would be crucial for understanding its function. The goal of this project is to utilize neural retina and RPE cells from control and JNCL patient-derived hiPSCs to delineate the role of CLN3 in the human retina. Important to the execution of this project and highlighting the role of CLN3 in hiPSC-derived retinal cell model, 1) robust expression of CLN3 protein is observed in hiPSC-derived retinal cells from control (unaffected subjects) (see image below) and 2) JNCL-patient-derived hiPSC-retinal cells display important pathological manifestations of the disease including increased autofluorescent material accumulation and presence of fingerprint inclusions within the cells, the two phenotypic hallmarks of the human disease.
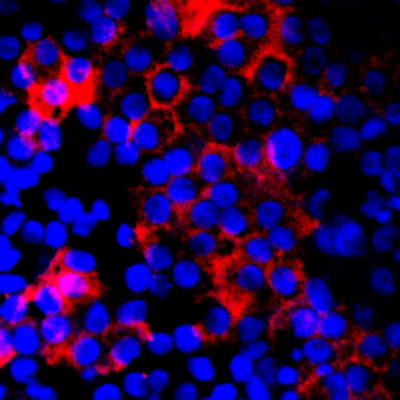
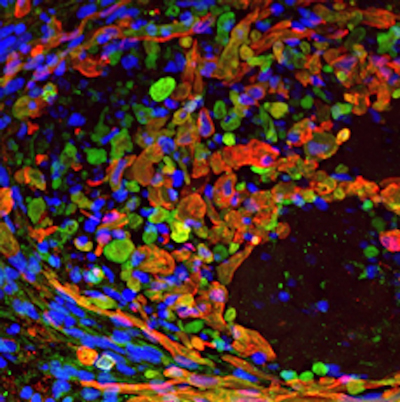
Representative confocal microscopy images showing CLN3 localization (red) in hiPSC-derived optic vesicles (left) and retinal pigment epithelium or RPE cells (right). Note: Strong co-localization of CLN3 protein is seen with photoreceptor marker , RCVRN, in hiPSC-derived optic vesicle cultures.