Research Projects
Human Microbiome Communities
Human life is dependent on a diverse community of symbiotic microbiota that have co-evolved with their human hosts to modulate crucial aspects of normal physiology, metabolism, immunity and neurologic function. We are engaged in multiple microbiome studies in areas of neonatal development, osteoarthritis, osteomyelitis, early childhood caries, atopic dermatitis and the tumoral microbiome of oral squamous cell carcinoma.
Neonatal microbiome and development
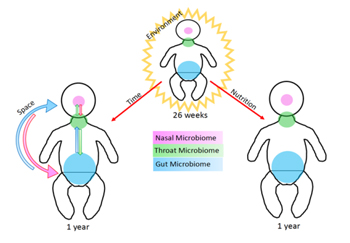
Coordinated development of pre- and full-term infant gut and respiratory microbiota through time and space
Our relationships with microbes begin in utero, with the limited microbial communities observed immediately after birth expanding into densely colonized, diverse bacterial ecosystems within the first weeks after birth. Early interactions that occur between members of the microbial community and between microbes and their human host are responsible for features of postnatal development that influence future health. The newborn infant microbiota is highly dynamic and undergoes rapid changes in composition through the first years of life towards a stable adult-like structure with distinct microbial communities of unique composition and functions at specific body sites. Relatively little has been reported about longitudinal microbiota development or compositional differentiation across multiple body sites during this period. This is particularly true for high-risk pre-term infants, who because of immature mucosal and skin barriers, as well as underdeveloped immunity and suboptimal nutrition, are at increased risk for invasive infection and dysregulated inflammation of critical systems, namely the respiratory and gastrointestinal tracts. Serious perinatal complications in these pre-term infants result in prolonged hospitalization, treatment with antibiotics and delays in enteral feeding that influence interactions with microbes and inhibit microbial colonization characteristic of full-term infants.
In ongoing studies of the gut and respiratory microbiome of 180 pre- and full-term infants from the multicenter Prematurity and Respiratory Outcomes Program (PROP) and the Respiratory Pathogens Research Center (RPRC) at the University of Rochester School of Medicine, we have demonstrated that: a) the neonatal gut and respiratory microbiota is shaped by innate and adaptive developmental responses and develops through a pattern of temporal progression through microbiota phases or community state types (CSTs), each differentiated by the abundance of specific taxa, b) temporal progression of the CST’s is influenced by infant maturity at birth and post-natal age, c) significant associations of microbiota across gut and respiratory body sites reveal distal connections and coordinated development of the infant microbial ecosystem, and d) there are significant associations between neonatal nutritional intake, phase of the gut microbiota, and preterm infant growth. Discovery of these relationships represent a significant first step in tailoring neonatal nutrient intake according to microbiota phase and establishment of clinical criteria for managing microbiota-based nutrient intake and care that supports optimal infant growth and development. The current studies are directed at understanding the relationships between respiratory and gut microbiota development and clinically significant respiratory outcomes. Our findings have revealed an unexpected relationship between respiratory disease and the gut microbiome, providing important insight into the gut-lung axis.
From the URMC Newsroom
Using the Microbiome to Help Premature Babies Grow
Role of the gut microbiome in initiation and progression of osteoarthritis
The obese microbiome is a key driver of osteoarthritis
Osteoarthritis (OA), a degenerative disease primarily affecting diarthrodial joints, afflicts 31 million people in the United States, with a global prevalence of disease recently estimated to exceed 250 million. The only clinically accepted treatment strategies are palliative, with no disease-modifying OA drug approved for use in humans. The growing incidence of osteoarthritis (OA) is linked to the expanding epidemic of obesity, with 66% of all OA patients classified as obese. While dogma has implicated joint overloading as causal, clinical and animal studies suggest that the comorbid association between OA and obesity includes a systemic inflammatory relationship, the nature of which is not defined. In a collaboration with Mike Zuscik and Bob Mooney, using a murine model of OA and obesity we discovered that the obesity-induced systemic inflammation and joint effects are associated with a proinflammatory gut microbiota. Treatment of the OA-obese mice with an insoluble prebiotic fiber that supports the expansion of Bifidobacterium pseudolongum and in turn production of gut small chain fatty acids (SCFA) shifted the microbiota to a non-inflammatory state and mitigated the obesity-related joint effects. Our current studies are directed at identification of causal relationships between OA and the gut microbiota and underlying immune mechanisms that moderate the systemic inflammation.
From the URMC Newsroom
The Bugs in Your Gut Could Make You Weak in the Knees
Type-2 diabetes and obesity associated immunodeficiency and the gut microbiome in osteomyelitis
Infection rates for osteomyelitis following total knee and hip arthroplasties are estimated to be around 1 to 3%, with Staphylococcus aureus the most frequent cause of these infections. Among the greatest risk factors for osteomyelitis are obesity and type 2 diabetes (T2D), as several studies have found that obese, diabetic patients are up to 5 times more likely to become infected following placement of an orthopedic device. Obesity and T2D (Ob/T2D) is associated with greater S. aureus infection severity, with increased colonization of bone and soft tissue, increased abscess formation and bone loss, and increased inflammation and macrophage abundance at the infection site. Similar to obese individuals with osteoarthritis, the gut microbiota in Ob/T2D individuals has an increased abundance of inflammatory taxa.
Based on these observations, we have initiated a new study with two major goals: 1) to determine the mechanistic basis of increased S. aureus virulence in the Ob/T2D environment and 2) to identify potential relationships between the inflammatory gut microbiota, host immunity and S. aureus virulence. Using a murine Ob/T2D orthopedic tibia implant infection model, we have demonstrated that reversal of the proinflammatory microbiome with insoluble prebiotic fiber and subsequent expansion of Bifidobacterium pseudolongum results in significantly reduced colonization of S. aureus in infected bone and soft tissue. Using an in vitro 3-D infection model, we have demonstrated increased expression of S. aureus adhesins (i.e. fibrinogen binding clumping factor A: clfA) in plasma from obese diabetic hosts relative to plasma from healthy individuals. We are using these models in our ongoing efforts to identify: a) functional changes in the Ob/T2D microbiota, b) innate and humoral immune mechanisms associated with shifts in the microbiota and reduction in S. aureus colonization, c) diabetic host factors that contribute to increased S. aureus virulence and d) adaptive responses of S. aureus to the Ob/T2D host environment.
Head and neck squamous cell carcinoma tumoral microbiome
Human microbiota colonizes mucosal surfaces on the skin, oropharynx, respiratory, digestive and urogenital tracts. Injury to these mucosal surfaces due to infection, trauma, and/or germline mutations can contribute to breach of mucosal barriers by the microbiota and development of cancer at these mucosal surfaces. Emerging clinical and molecular analyses have revealed an enrichment of specific microbial species in head and neck squamous cell carcinoma (HNSCC), colorectal, pancreatic, and other solid tumor cancers compared with adjacent normal tissue. In this study, we are using integrated genomic and immune approaches to examine mechanistic associations between the tumoral microbiota and the tumor microenvironment of HNSCC.
In our initial efforts, we evaluated the tumoral and microbiota transcriptomic signatures of paired normal and tumor samples from nine patients. Functional pathway analysis revealed a conserved cancer signature implicating enriched cancer, immune, and microbial related pathways and suggests the immune microenvironment is differentially regulated in cancer. Taxonomic 16S rRNA analysis of the paired samples demonstrated an increased abundance of both Fusobacterium nucleatum and Capnocytophaga ochracea, as well as a reduction in Streptococcus mitis abundance in tumor tissue relative to normal. Metatranscriptome analysis identified gene families and pathways associated with F. nucleatum that may contribute to or enable tumor growth. To assess potential microbiome and immune heterogeneity in the HNSCC tumoral tissue, we performed simultaneous analyses on 1 normal region and 6 malignant regions from a single HNSCC resection. Hierarchical all-against-all significance testing was used to identify bacteria that co-varied with suppressive immune cell populations. Significant associations between inflammatory monocytes and the Neisseria eikenalla and Haemophilus influenza were observed across tumor locations, suggesting their role in promoting the suppressive phenotype of intratumoral monocytes.
The early life oral microbiome and early childhood caries
Early childhood caries (ECC), the single most common chronic childhood disease, disproportionately afflicts up to 72.7% of underprivileged preschool children in both developing and industrialized countries. This disease substantially/adversely impacts children, families, and public health systems. Dental caries is a communicable, biofilm (plaque)-dependent infectious disease. Traditional microbial risk markers for ECC include Streptococcus mutans and Lactobacillus species. Significantly, in the past decade, clinical studies have also observed that Candida albicans is highly abundant in the oral cavity of children with ECC, together with S. mutans. In collaboration with the Eastman Dental Institute (Dorota Kopycka-Kedzierawski and Jin Xiao), the Center for Oral Biology (Robert Quivey) and Department of Psychiatry (Tom O’Connor), we have undertaken a prospective cohort study to investigate the prevalence of oral carriage of C. albicans in infants at high risk for S-ECC and their mothers, the association between oral C. albicans carriage and onset of S-ECC, and interactions between oral C. albicans and S. mutans in early life. A central aspect of this study is development of the oral microbiota in early life prior to and following tooth eruption and the contribution of maternal oral microbiota to ECC.
Atopic Dermatitis
Atopic Dermatitis (AD) is the most common inflammatory skin disease, affecting 15 million Americans (17% of children and 10% adults) with most severe disease seen in adult patients. The impact of AD on quality-of-life is greater than any other chronic skin condition. AD is characterized as a common, chronic pruritic, inflammatory skin disease complicated by recurrent infections of Staphylococcus aureus, which occur in 55%-90% of AD patients. The mechanism underlying persistent bacterial colonization in AD is poorly understood. An increase in the proportion of Staphylococcus spp. and a reduction in diversity of the skin microbiota is associated with more severe disease. In collaboration with the Atopic Dermatitis Research Network (ADRN) and Lisa Beck (URMC Dermatology), we are participating in an interventional clinical trial with Dupilumab (a humanized monoclonal antibody directed against the IL-4 receptor alpha subunit (IL-4Rα), for the treatment of adults with moderate-to-severe AD who are not adequately controlled with topical prescription therapy. In this study, outcome measures include changes in AD skin microbiota function and host immune responses following treatment with Dupilumab and integration of microbiota function clinical outcomes.
Pathogenesis and Genomics of Bacterial Pathogens
Staphylococcus aureus and Clostridioides difficile are among the most successful of human pathogens, capable of causing invasive and deadly opportunistic infections. We are using in vivo, biochemical and genomic approaches to identify genetic pathways and mechanisms associated with S. aureus responses to host innate immunity and adaptation of the clonal complex 30 lineage to a persistent lifestyle in the host. In addition, we are using genomic and phylogenetic approaches to assess emergence and transmission of new S. aureus and C. difficile clinical isolates throughout hospitals, long-term care facilities and the general community in Monroe County NY.
Staphylococcus aureus pathogenesis
Staphylococcus aureus is an opportunistic pathogen that causes a wide range of clinical pathology and disease, ranging from asymptomatic skin colonization to death. It adapts to extremely diverse environments within the human host, both as an extracellular pathogen and as an invasive pathogen of non-phagocytic cells. It infects effectively all organ systems, encompassing countless metabolic niches. S. aureus causes purulent infections, such as styes, impetigo, osteomyelitis, and infective endocarditis (IE), as well as nonpyogenic disease via toxins, such as Staphylococcal food poisoning and toxic shock syndrome. S. aureus is a common cause of the rare pericarditis and myocarditis, as well as IE. The clinical presentation of disease is reflected in the genomic, transcriptomic, and proteomic character of a given strain of S. aureus.
The public health concern caused by S. aureus is truly immense. Bacteremia caused by the organism is fatal in as many as 30% of cases, and leads to more deaths than AIDS, tuberculosis, and viral hepatitis combined. Nasal carriage is the primary reservoir of S. aureus, and in industrialized countries colonization is approximately 30%. Of those colonized, carriage at distal body surfaces is increased dramatically; these body sites serve as secondary reservoirs of infection and increase risk of systemic infection. Compared to other developed nations such as Canada, the United States has double the incidence rate of bacteremia and increased prevalence of methicillin-resistant Staphylococcus aureus (MRSA). S. aureus infections are dichotomized into two groups: healthcare associated infections (HA) and community associated infections (CA). Historically, most MRSA infections were healthcare acquired and affected the immunocompromised and elderly, though increased rates of CA-MRSA have been observed in recent years and lines between healthcare and community associated S. aureus have blurred significantly. Financially, the annual cost to society from CA-MRSA in the United States alone is as high as $13.8 billion, a figure that does not account for the myriad other methicillin sensitive strain of S. aureus or traditional HA-MRSA.
Our current studies are focused on S. aureus isolates in the contemporary clonal complex 30 (CC30) lineage. S. aureus isolates in this lineage are genetically more divergent than other clonal complexes, are associated with persistence at the mucosa and appear to have adapted to a more persistent, less virulent infection pattern. Genomic analyses of CC30 revealed numerous pseudogenes and horizontally acquired genes mobile genetic elements that may contribute to the ability of this lineage to survive in this unique niche. Two of the unique elements are a putative nitric oxide reductase and a novel global regulator (MocR) that participate in bacterial responses to host innate immunity and adaptation to a persistent lifestyle in the host.
a) Nitric oxide reductase. The production of nitric oxide (NO) by activated phagocytes is a major host response to which S. aureus metabolically adapts through multiple strategies that are conserved in all CCs, including an S. aureus nitric oxide synthase (Nos). Previous genome analysis of CC30, a lineage associated with chronic endocardial and osteoarticular infections, revealed a putative NO reductase (Nor) not found in other CCs that potentially contributes to NO resistance and clinical outcome. We have demonstrated that Nor has true nitric oxide reductase activity, with nor expression enhanced by NO stress and anaerobic growth. Furthermore, we demonstrated that nor is regulated by MgrA and SrrAB, which modulate S. aureus virulence and hypoxic response. Transcriptome analysis of S. aureus UAMS-1, UAMS-1 Δnor, and UAMS-1 Δnos under NO stress and anaerobic growth demonstrated that Nor contributes to nucleotide metabolism and Nos to glycolysis. We demonstrated that Nor and Nos contribute to enhanced survival in the presence of human PMNs, have organ specific seeding in a tail vein infection model and that Nor contributes to abscess formation in an osteological implant model. We also demonstrated that Nor has a role in S. aureus metabolism and virulence. The regulation overlap between Nor and Nos point to an intriguing link between regulation of intracellular NO, metabolic adaptation, and persistence in the CC30 lineage.
b) MocR. The ability of S. aureus to adapt to a diverse range of infection sites and host environments is key to its success as a pathogen. We have demonstrated that global regulation by MocR of pathways that contribute to amino acid and carbohydrate metabolism is essential to the ability of S. aureus CC30 in facilitating nutrient acquisition and competing with host microbiota for host nutrients. We identified a potential role for MocR in the regulation of multiple amino acid transport and metabolism pathways and co-regulation of branched chain amino acids (BCAA) by MocR and CodY, suggesting a novel regulatory mechanism of metabolic adaptation in the CC30 lineage. Our results indicate that MocR and CodY may work synergistically to regulate nutrient stress survival genes. CodY has been reported in many different Gram-positive strains and S. aureus backgrounds to regulate genes required for nutrient biosynthesis and stress in times of nutrient depletion. The additional layer of regulation by MocR, may provide an advantage to CC30 strains, allowing them to outcompete other bacterial species. Our results suggest that the MocR regulatory mechanism is complex as MocR also regulates expression of other S. aureus regulators, including DeoR, GntR, and MalR. Our current efforts are focused on evaluating the regulatory mechanism of MocR and it’s role in adaptation of S. aureus CC30 to a persistent lifestyle.
Genomics of Staphylococcus aureus and Clostridioides difficile
The bacterial pathogens, Clostridioides difficile and Staphylococcus aureus are major public health threats, with infections associated with hospital settings, long-term care facilities and the general community. Developing and implementing effective infection control measures requires an understanding of C. difficile and S. aureus transmission dynamics, including the movement of patients across the healthcare and the community settings, and identifying potential reservoirs of these pathogens. In collaboration with the Centers for Disease Control, infectious disease clinicians at URMC (Ghinwa Dumyati and Nicole Pecora) and colleagues in the Department of Microbiology (Michelle Dziejman), we are sequencing the genomes of S. aureus and C. difficile isolates collected from patients at the major clinical centers (URMC and RGH) and long-term care facilities throughout Monroe County NY. Phylogenetic analysis of the whole genome sequence data is being integrated with epidemiological surveillance data to develop a spatial-temporal analysis of transmission patterns between the clinical and long-term care facilities and the community. This approach has the potential to expand our understanding of these two pathogens and guide healthcare and community policies to track strain prevalence and infection.