Algorithmic analysis and exploration of high-parameter flow cytometry data
SWIFT automated clustering and sample comparison
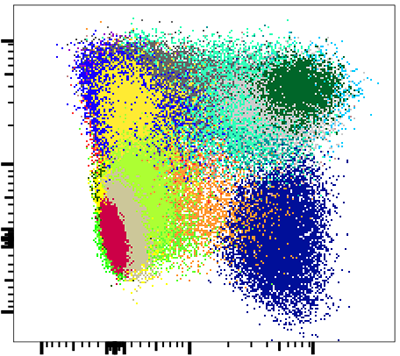
SWIFT clusters
Manual analysis of high-dimensional flow cytometry data is a subjective, unreproducible technique, and exhaustive manual analysis of high-dimensional flow data is not feasible. We have developed a high-resolution, model-based flow cytometry data clustering program, SWIFT, that has higher resolution than most other gating or clustering algorithms, and detects sub-populations at levels as low as one part per million. SWIFT is particularly good at finding rare populations, and has been extensively validated for finding rare cytokine-expressing T cells in antigen-stimulated PBMC.
SWIFT initially uses an iterative weighted sampling technique to fit the data to a large number of Gaussian distributions, then further splits these populations until all are unimodal in all dimensions. Overlapping sub-populations are then merged to arrive at an objective estimate of both the number and shape of all cell populations in the sample.
Sample comparison using cluster templates
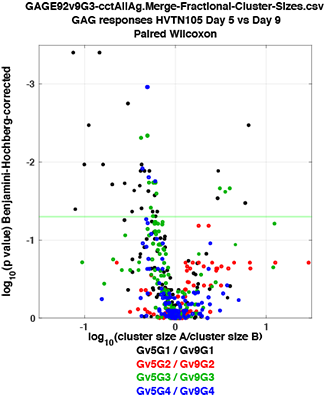
Rigorous comparison of clusters affected by vaccination
To compare samples, a SWIFT cluster template is generated from e.g. a consensus sample, and all cells in additional samples are assigned to this template. This facilitates rigorous comparison of large numbers (thousands) of samples, and enumerates small sub-populations, even 0 cells in a negative control. We enhanced the templating approach with a cluster template competition method that sharpens the detection of differences between biological groups, e.g. young vs elderly.
SWIFT is a set of MATLAB(r) programs
Registration of data between batches
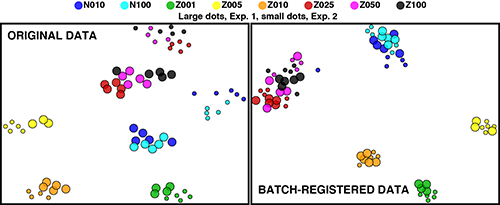
t-SNE representation of raw and registered data. Collaboration with Rusty Elliott
To combat batch effects that can occur due to day-to-day variation in large studies, even with rigorous quality control methods, we developed a novel fully-automated registration approach. A SWIFT cluster template is produced from a concatenate (or control sample) from a reference batch, and other batch concatenates are assigned to this template. The new channel medians for each cluster are used to calculate the shifts required to register each cluster to the reference batch. These shifts are then applied to individual samples. As the shifts are calculated using batch concatenates, differences between biological groups are not affected. Thus batch registration has the remarkable property of decreasing batch variation, while normally enhancing the resolution of biological groups.
Making exploration of complex data fast and easy
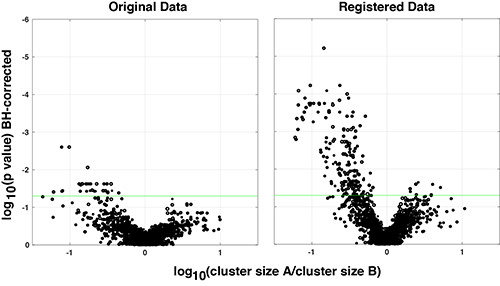
Registration improves the ability to identify biological differences
Because SWIFT clustering is not influenced by operator pre-conceptions regarding population definitions, this algorithm often identifies unsuspected sub-populations, making it an ideal tool for data exploration. A suite of pre- and post-processing tools has been developed around SWIFT clustering, facilitating data curation, extensive QC tests, and exploration and visualization of data in many interactive graphical and tabular outputs.
SWIFT, Registration and associated exploration tools are currently being added to the Galaxy interface of the IMMPORT database to make these tools generally available (supported by grant UH2 AI132339).
All the images on this page were produced directly by these exploration tools. See also Projects 2 and 3 for additional examples of the output of our tools.
References
- Naim I, Datta S, Rebhahn J, Cavenaugh JS, Mosmann TR, Sharma G. SWIFT-scalable clustering for automated identification of rare cell populations in large, high-dimensional flow cytometry datasets, Part 1: Algorithm design. Cytometry A. 2014;85(5):408-21. doi: 10.1002/cyto.a.22446. PubMed PMID: 24677621.
- Mosmann TR, Naim I, Rebhahn J, Datta S, Cavenaugh JS, Weaver JM, Sharma G. SWIFT-scalable clustering for automated identification of rare cell populations in large, high-dimensional flow cytometry datasets, Part 2: Biological evaluation. Cytometry A. 2014;85(5):422-33. doi: 10.1002/cyto.a.22445. PubMed PMID: 24532172.
- Aghaeepour N, Finak G, The FlowCAP Consortium, The DREAM Consortium, Hoos H, Mosmann TR, Brinkman R, Gottardo R, Scheuermann RH. Critical assessment of automated flow cytometry data analysis techniques. Nat Methods. 2013;10(3):228-38. Epub 2013/02/12. doi: 10.1038/nmeth.2365. PubMed PMID: 23396282.
- Rebhahn JA, Roumanes DR, Qi Y, Khan A, Thakar J, Rosenberg A, Lee FE, Quataert SA, Sharma G, Mosmann TR. Competitive SWIFT cluster templates enhance detection of aging changes. Cytometry A. 2016;89(1):59-70. doi: 10.1002/cyto.a.22740. PubMed PMID: 26441030.