Role of Interleukin-1 in Alzheimer’s Disease Neuropathogenesis
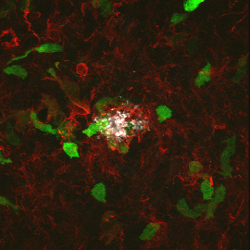
Peripheral infiltrating cells (green) in vicinity of an amyloid
plaque surrounded by iba positive microglia (red).
Alzheimer’s disease is the leading cause of dementia in our aging population, affecting as many as 40% of people over the age of 85. Morphological changes in microglia and astrocytes known as activation, as well as local production of proinflammatory cytokines, chemokines, and complement components were described over 20 years ago. Together with data that nonsteroidal anti-inflammatory drugs appeared to lower the risk of AD, these changes have spurred great interest in understanding how neuroinflammation contributes to disease pathogenesis. Our current studies supported by the National Institute of Aging focus on the role of interleukin-1 (IL-1), a key proinflammatory cytokine, in modulating pathological hallmarks of AD, including both amyloid plaques and neurofibrillary tangles.
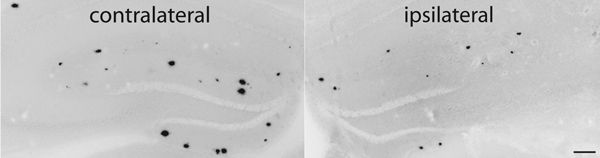
Reduction of amyloid plaque burden (ipsilateral image) following IL-1 overexpression.1
As one of the proinflammatory cytokines shown to be elevated in AD brain, IL-1 was believed to take part in a vicious cycle of pathology and cytokine production by microglia responding to amyloid plaque deposition. To investigate this further, we created a transgenic mouse in which we could induce sustained IL-1 expression in a regionally and temporally specific manner (IL-1XAT). Much to our surprise, when these mice were crossed with a mouse model of Alzheimer’s disease, we found that IL-1 expression led to a reduction in amyloid plaques, rather than the anticipated exacerbation of pathology. Our working hypothesis is that the local inflammation induced by IL-1 enhances phagocytosis, perhaps by infiltrating macrophages, of extracellular amyloid. To explore this hypothesis we are making use of mice lacking chemokine receptors required for macrophage recruitment as well as chimeric mice harboring bone marrow.
We are also using IL-1XAT mice to study the influence of neuroinflammation on tau pathology. For these studies we have crossed IL-1XAT mice with the P301S tau mouse model, which develops neurofibrillary tangles. Based on work in other laboratories we anticipate that IL-1 will exacerbate tau phosphorylation through the activation of specific protein kinases.
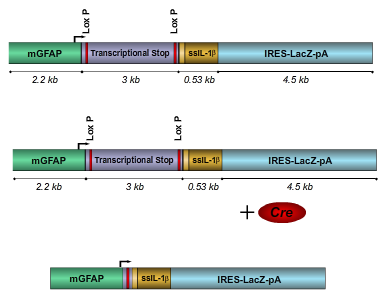
Schematic of IL-1β XAT transgene. Introduction of Cre recombinase
results in transcriptional activation of human IL-1β.1
Finally, we have initiated additional studies to explore the effects of neuroinflammation driven by IL-1 on hippocampal dependent learning and behavior. Our preliminary results show significant decline in several behavioral tasks, including contextual fear conditioning and the Morris water maze. Understanding molecular and electrophysiological correlates of these effects represents a new area of exploration in our laboratories.
Footnotes
- 1
Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O'Banion MK (2007 Jun 05). Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 117, 1595-604.