CART Newsletters
CART Newsletters
Fall 2025 Content
Easy One-Pot Moron Pulao (Bangladeshi Chicken & Rice Dish)
Contributed by Tamjeed Sayeed Kashem
This traditional Bangladeshi dish blends aromatic spices and tender chicken with flavorful Basmati rice, all cooked effortlessly in one pot.
Ingredients:
For the Chicken & Marinade
- Chicken: 1 kg whole chicken, cut into large pieces.
- Marinade: 1 tsp Salt, ½ tsp Red chili powder, 1 tsp Lime juice, ½ tsp Ginger paste.
Aromatics & Spices
- Pastes: 3 tbsp Onion paste, 1 tbsp Garlic paste, ½ tsp Ginger paste, 4 tbsp Plain yogurt, 1 tbsp Cashew paste, 1 tsp Poppy seed paste, 1 tsp Green chili paste.
- Whole Spices: 2 Bay leaves, 3 small Cinnamon sticks, 5 Cardamom pods, 7-8 Cloves.
- Spice Mix (Morog Pulao Spice Mix): 1 blade mace, ½ nutmeg, ½ tsp white pepper, ½ tsp cumin, ½ tsp garam masala.
Other Gravy Ingredients
- 3-4 tbsp Cooking oil, 1 tbsp Unsalted butter, 3 tbsp Sliced onion.
- 7-8 Green chilies, 1 tsp Sugar, 1½ tbsp Raisins, 1 tbsp Tamarind pulp.
For the Pulao Rice
- Rice: 3 cups Kalijeera or Basmati rice, soaked for 30 min.
- Liquid: 4½ cups Hot water, 1 cup Warm milk.
- Flavorings: 4-5 Aloo bukhara (dried plums), ½ tsp Kewra water, ½ tsp Rose water.
- Garnish & Finishing: 4 Boiled eggs, ¾ cup Fried onions, crushed, 1 tbsp Ghee.
Instructions:
1. Marinate and Fry the Chicken
- Marinate: Mix the chicken pieces with 1 tsp salt, red chili powder, lime juice, and ginger paste. Let it marinate for 30 minutes.
- Fry: Heat the cooking oil and butter in a large pan. Fry the marinated chicken pieces in batches for 2–3 minutes until they color, then set them aside.
2. Cook the Spice Base
- Sauté Aromatics: In the same pan, add the bay leaves, cinnamon, cardamom, cloves, and sliced onions. Cook until the onions are lightly browned.
- Add Pastes: Stir in the onion, garlic, and ginger pastes. Add a splash of water and cook until the raw smell disappears.
- Combine and Simmer: Return the chicken to the pan. Add the yogurt, tamarind pulp, green chilies, sugar, cashew and poppy seed pastes, and the Morog Pulao spice mix. Mix well.
- Finish Chicken Gravy: Stir in the fried onions, raisins, milk, and green chili paste. Cook covered on medium heat for 20 minutes, stirring occasionally.
3. Cook the Pulao Rice
- Remove Chicken: Take the cooked chicken pieces out of the pan and set them aside.
- Prepare Rice: Add the soaked rice to the pan and stir it in the chicken gravy for 2–3 minutes.
- Add Liquid & Flavor: Add the hot water, additional salt, aloo bukhara, kewra water, and rose water.
- Cook Rice: Once the liquid is simmering, cover the pot and cook on low heat for 15–20 minutes.
4. Final Layering and "Dum" (Steam)
- Layer: Gently make space in the center of the rice and add back the chicken pieces and boiled eggs.
- Garnish: Cover the chicken with the rice, sprinkle with more fried onions, and drizzle with ghee.
- Final Cook: Cover the pot and cook on the lowest heat for a final 10 minutes (on dum).
Serve hot and enjoy!
Summer 2025 Content
Bang Bang Chicken Skewers
Contributed by Meghann O'Brien
Ingredients
- 2 chicken breasts cut into 1-inch cubes
- 3 tablespoons creamy peanut butter
- 2 tablespoons sriracha
- 1 tablespoon honey
- 2 cloves minced garlic
- 2 tablespoons soy sauce or tamari (if gluten-free)
- Juice of 1 lime
- Wooden skewers soaked in water for 30 minutes
Instructions
- In a bowl, whisk together peanut butter, sriracha, honey, garlic, soy sauce, and lime juice to make the marinade.
- Toss chicken cubes in half the marinade and refrigerate for 30 minutes to 4 hours.
- Thread chicken pieces onto soaked wooden skewers.
- Cook skewers by grilling, cooking in an air fryer at 400°F (200°C) for 10-12 minutes, turning halfway; or baking in oven at 425°F (220°C) for 15-18 minutes, flipping once.
- Brush skewers with remaining marinade in last 5 minutes of cooking.
- Ensure chicken is cooked through (internal temperature 165°F/74°C), then let rest 3-5 minutes before serving.
- Enjoy!
Winter 2025 Content
Meet Ravi Misra
Q&A with the new Associate Scientific Director of the Flow Cytometry Resource
What initially sparked your curiosity about biology and research? Was there a defining moment or experience?
While taking Advanced Biology in high school, I distinctly recall learning about the transmission of genetic diseases through generations. What fascinated me even more was how, at the time, genetic engineering was a brand-new concept that could be developed into a revolutionary technology with the potential for treating genetic diseases. At the same time, a stone’s throw from my high school the biotechnology sector in Cambridge MA was growing. The event that cemented my decision to study biomedical sciences as my career, came when I attended a program for high school students during a spring break held at the Whitehead Institute. Hearing world renowned scientists present their work, and touring the research labs, left an indelible mark on me. As an undergraduate, I took courses in cell/molecular biology and immunology, which piqued my interest in studying how the complex immune system must respond to infectious diseases and then turn off, only to be turned back on in response to a subsequent infection. So, I knew at a young age that I wanted to become a scientist.
You’ve been with the institution for several years. How has your work in the Pryhuber Lab shaped your scientific perspective?
During my graduate studies and in my first postdoctoral training period, I learned methods for utilizing flow cytometry to study cells of the immune system. This ranged from elucidating T cell signaling cascades to studying B cell responses in influenza infection and how deletion of a G-protein leads to an autoimmune syndrome in mice. As I transitioned into the Pryhuber lab, I was responsible for developing methods to sort primary human immune cells from cord blood and pediatric peripheral blood, which marked a distinct career right turn to studying human sample.
Serendipity struck in 2013 when Dr. Pryhuber competed for a slot on the LungMAP program. I was able to dig out my knowledge of lung biology gained as a graduate school trainee at UVM who was being supported by an NIH-sponsored institutional training grant. I spearheaded efforts to establish methods for isolating cells from primary human lung samples, which were used to win LungMAP grant funding as the Human Tissue Core (HTC). This method has been used to generate RNASeq and unbiased proteomics datasets. We are currently serving as the LungMAP HTC through the sunset of the program in 2028. I also serve as the Program Manager for the HuBMAP-Lung project, where I am responsible for coordinating research efforts across four research centers as we build a multimodal map of normal human lung. In addition to those projects, I have also worked on several projects involving human subjects, which has given me new perspectives in designing protocols that fall under the purview of the Institutional Review Board. Overall, the work in the Pryhuber lab has expanded my skill set to include studying human cells at cellular and molecular levels.
What do you find most fascinating about flow cytometry as a technology?
I find that extensive cellular phenotyping using flow cytometry, CyTOF, and imaging cytometry allows for an in-depth study of how cell populations change in disease states and during normal postnatal human development. Importantly, studies can be completed on cells from just about any lineage, which has cemented flow cytometry as an important tool used in biomedical research beyond studying the immune system. By using spectral flow cytometry, limitations posed by cellular autofluorescence, and spectral overlap of fluorophores can be overcome to sort six cell populations which increases the material that one can isolate in a single sort run versus the old instruments. Additionally, the vast range of downstream analyses that can be performed on cells isolated by sorting is very powerful.
What are some innovations or goals you hope to bring to the facility in the coming years?
I envision more labs using flow cytometry and other instrumentation in the core to study their cells of interest and advance their research programs. For instance, spectral flow has increased the number of analytes that a researcher can study at one time. Additionally, spectral flow helps to account for autofluorescence as it can mask signal that has been a big problem for studying cells isolated from the lung. This is an example of how I can use my experience in studying cells isolated from complex tissue digests to help other investigators. I would also like to help new investigators who are starting labs to learn how flow cytometry could help advance their research program. This would include planning rigorous experiments for testing hypotheses and ensuring that their lab personnel will be successful in completing the experiments. Finally, I would love to leverage the resources of the core and the vast research enterprise at URMC to expand work with clinical trials. We are well positioned to build a pipeline for winning additional funding from the NIH and the private sector that would help to improve human health.
Do you have a favorite book, mentor, or figure in science who has inspired your work?
This is a complicated question. I would say that there are too many mentors to list. Mentors from high school, in college, through graduate school, into my postdocs, through my time as a faculty member have provided guidance and advice that shaped who I am today. I feel strongly that paying the deed forward and mentoring the next generation of biomedical researchers is critical, and I try to support training the next generation of scientists to the best of my ability.
Spicy Beef Queso
Contributed by Meghann O'Brien
Ingredients
- 1 pound ground beef
- 2 jalapenos, finely minced
- 32 ounces Velveeta cheese, cut into cubes
- 2 cans of Rotel
Instructions
- In a large skillet, brown the ground beef over medium heat until the meat is no longer pink. Drain.
- In a 4 to 6-quart slow cooker, add the prepared beef mixture, the Velveeta cheese and the cans of Rotel and minced jalapenos.
- Stir well and cook on low until the cheese is completely melted, 1 to 2 hours. Keep the slow cooker set to warm to serve during a party, giving the mixture a good stir before serving.
- Serve with tortilla chips, Fritos Scoops, pretzels, or cut vegetables.
Fall 2024 Content
Pumpkin Soup Recipe from the Moosewood Restaurant
Contributed by Tim Bushnell, PhD
As fall arrives, bringing with it cooler weather, it’s the perfect time to indulge in classic comfort foods like chili, soups, and stews. One of my favorite seasonal ingredients is squash. With such a wide variety available, squash is incredibly versatile, from the iconic pumpkin pie to savory dishes like spaghetti squash Bolognese.
There’s something particularly satisfying about a warm bowl of pumpkin soup with freshly baked bread on a chilly night. One recipe I love comes from the Moosewood Restaurant in Ithaca. Here’s how to make it:
Moosewood Pumpkin Soup Recipe:
Ingredients:
- 2 cups chopped onions
- 2 tablespoons olive oil
- 1/2 cup carrots, peeled and sliced
- 1/2 cup parsnips, peeled and sliced
- 1 1/2 teaspoons salt
- 2 1/2 cups light vegetable stock
- 1 1/4 cups unsweetened apple juice
- 1/2 cup tomato juice
- 1 teaspoon ground cumin
- 1/2 teaspoon ground nutmeg
- 1/2 teaspoon ground cinnamon
- 1/2 teaspoon paprika
- 1 3/4 cups cooked pumpkin (~15 oz can of pumpkin puree)
Instructions:
1. In a soup pot, sauté the onions in olive oil until translucent, about 10 minutes.
2. Add the carrots, parsnips, and salt, and continue sautéing for another 5 minutes.
3. Stir in the vegetable stock, apple juice, tomato juice, cumin, nutmeg, cinnamon, and paprika. Cover and bring to a boil, then reduce the heat and simmer until the vegetables are tender.
4. Stir in the cooked pumpkin.
5. Use an immersion blender to puree the soup to your preferred texture, whether smooth or slightly chunky.
For a richer flavor, I recommend using fresh pumpkin. To prepare it, cut a pumpkin into sections, coat lightly with oil (olive or avocado), and season with salt and pepper. Roast the pieces in a 400°F oven for about 30 minutes. If you enjoy garlic, consider roasting a few cloves alongside the pumpkin for added depth.
And for some added crunch, make your own roasted pumpkin seeds. After you wash and dry the pumpkin seeds, place them in a bowl and coat with some olive oil. Season with salt, pepper, and garlic powder to taste. Bake in a 350F oven for about 15 minutes. Stir the seeds every 5 minutes or so to ensure even roasting. Let them cool and top your soup with these seeds.
Enjoy this heartwarming soup and the cozy flavors of fall!
Summer 2024 Content
The MSRL Leads New York in Proteomics
by Kyle Swovick, PhD
Within the past 8 months, the Mass Spectrometry Resource Laboratory (MSRL) has been working to completely overhaul instrumentation and services they provide. Now housed within the MSRL are two brand new, state-of-the-art mass spectrometers; the Orbitrap Astral by Thermo Fisher and the timsTOF Ultra by Bruker Daltonics (soon to be upgraded to the Ultra 2 which was released in June). As the only lab in New York to house both instruments— and one of the few in the country the acquisition of these two instruments positions researchers at the University of Rochester to performing cutting-edge proteomic experiments. Due to the significantly enhanced capabilities of these instruments, the MSRL has also actively been developing several exciting techniques for researchers to take advantage of including label-free phosphorylation enrichment, extracellular vesicle (EV) enrichment from plasma, immunopeptidomics, and single cell proteomics. While the Astral and Ultra will both be dedicated to proteomic experiments, they’re unique designs offer distinct advantages that will allow them to excel at specific experiments.
The Astral, which replaced our previous proteomic mass spectrometer, the Fusion Lumos, is a unique tribrid instrument that combines an Orbitrap mass analyzer with a time-of-flight (TOF) mass analyzer. As a result, it has both the unrivaled mass resolution of an Orbitrap and the blazingly fast scan speeds of a TOF. On top of this, Thermo Fisher’s unique spin on the traditional TOF design (ASymmetrical TRAck Lossless, Astral) results in higher sensitivity and resolution than traditional TOFs. Functionally, this means proteomes can be quantified at a greater depth than before while simultaneously decreasing the time needed to analyze each sample (Table 1). The combination of high resolution, sensitivity, and scan speeds means the Astral will shine brightest in proteomics experiments when there is a wide dynamic range of protein abundances (i.e., plasma) or large number of proteins being expressed. Therefore, any global proteomics experiments, whether from cell lines, tissue, or plasma will be analyzed with the Astral. While the Astral is an amazing advancement for LC-MS/MS-based proteomics, its biggest limitation is the inability to perform multiplexing with tandem mass tags (TMT), which was our primary method for performing phosphorylated peptide enrichment. As a result, the acquisition of the Astral necessitated a new method for enriching and analyzing phospho peptides which is now done through the use of MagResyn’s Ti-IMAC beads. This new method allows the MSRL to enrich phospho peptides from as little as 25µg of digested peptides instead of the 100µg we previously needed for TMT labeling. This means less starting material required for the researcher to prepare for each replicate. Because of this method, we are now also able to analyze these enriched samples using data-independent acquisition (DIA, see previous newsletters for greater detail), resulting in increased coverage and decreased ratio compression.
The timsTOF Ultra (and Ultra 2) is a variation on the timsTOF line of hybrid mass spectrometers developed by Bruker for the past decade that boast a powerful TOF mass analyzer prefaced by a TIMS (Trapped Ion Mobility Spectroscopy) device. The addition of TIMS before the mass spectrometer offers two distinct benefits: 1) all non-tryptic peptides have a different mobility than tryptic peptides so they can be filtered out of the analysis before even entering the mass spectrometer and 2) it can accumulate tryptic peptides before they enter the mass spectrometer. By combining these two features, along with unique hardware and electronics developed for the Ultra and Ultra 2, the MSRL is now able to perform proteomic experiments that require sensitivity that was unimaginable until now including the rapidly growing field of single cell proteomics (SCP) along with immunopeptidomics. We are actively developing SCP methodologies so they can be applied broadly to a wide range of cell types and tissues. Just as a teaser, we recently were able to quantify nearly 2000 proteins and 6000 peptides from a single HeLa cell using the Astral. While the coverage varied between various single cell runs, we believe that with further optimization of our isolation, lysis, and digestion protocols, along with analyzing the digests using the Ultra 2, our coverage and consistency will dramatically improve. Please stay tuned for future updates from the MSRL for when we are ready to fully offer SCP services to the research community and see how it can bring your research to a whole new level.
These two instruments place the University of Rochester at the forefront of proteomic research and the types of analyses now available to researchers here are unparalleled. If you want to learn more about one or both of these instruments or how they can help you answer your unique biological questions, please contact anyone from the MSRL.
The FCR Introduces New Cell Sorters
by Matt Cochran
At the end of 2024 we will sadly have to say goodbye to our FACSAriaII cell sorters. Last year we also had to retire our BD LSRII analytical flow cytometers, so there’s been a large amount of “forced” instrument turnover recently. Unfortunately, due to end of service decisions by BD and the workhorse nature of the sorters in general we can no longer support the use of the Arias after 12/31/2024.
However, with these “forced” changes we also have the opportunity to bring in new exciting technologies, and expand the capabilities we, as a shared resource laboratory, can offer our investigators. This worked out beautifully in the case of the BD Symphony A1, and Cytek Aurora instruments when the LSRIIs were winding down. The A1s are direct replacements for the LSRs and have been integrated pretty seamlessly with our user base. The Cytek Aurora analyzers provided a new option for analytical flow with new capabilities and have proven to be excellent tools with a growing group of excited investigators.
For cell sorting we’ve also brought in two new options that will allow us to provide similar services and also build on our capabilities as well. The first new option from last year was the Cytek Aurora CS (Muppet Name: Link Hogthrob), and we’ve been very happy with its performance. We also just installed a new BD S6 (Muppet name under development) cell sorter and look forward to getting to put it through its paces. The anticipated opening date for the S6 is August 1st but watch the UR_Cytometry email list for more information.
Below is a short list of highlights for each of our new sorters:
Aurora CS (Link Hogthrob) Highlights:
- Simultaneous collection of up to 6 populations.
- Full spectrum flow with 5 lasers – matched configuration with the 5 laser analyzer (Sweetums).
- Direct analyzer communication and transfer – experiments tested and files/gating strategies created on Sweetums can be imported into the CS, dramatically decreasing the setup and development time for your sorting projects.
- Autofluorescence extraction can improve resolution in some tricky samples. Check this paper for details: DOI: 10.1002/cyto.a.24885
- Excellent longitudinal stability for consistent fluorescence and scatter signals
- No need to re-run unmixing controls as long as experimental conditions are not changed, again reducing setup and sample prep time.
BD S6 Highlights:
- Simultaneous collection of up to 6 populations.
- Traditional flow with 5 lasers and 24 fluorescence detectors (includes a UV laser).
- Similar (but not matched) to the LSR Fortessa (Dr. Teeth and Camilla) with 6 detectors off the UV laser.
- Updated version of the FACSAria.
- Runs using FACSDiva software which is familiar to many investigators.
- Instrument hardware is updated but similar to the Aria’s which increases familiarity and assists with troubleshooting.
In closing, if you or your lab are eager to advance your research with these new tools or any other equipment and capabilities within the FCR, we are here to support your efforts. We would love to have an in-depth discussion to help you maximize your lab’s potential. Please reach out to us at flowcytometry.urmc.edu, and together, we can achieve great results!
Italian-Style Spumoni Ice Cream Cake with Amaretti Cookies
by Megan Crawford
Ingredients:
- 1 quart pistachio ice cream, softened
- 1 quart cherry ice cream, softened
- 1 quart chocolate ice cream, softened
- 1 cup chopped maraschino cherries, drained
- 1 cup chopped pistachios
- 1 cup chocolate fudge sauce
- 1 cup whipped cream
- 1 package amaretti cookies, roughly crumbled
- Optional: Additional maraschino cherries and rainbow sprinkles
Instructions:
Prepare the Pan:
- Line a springform pan (9 inches) with plastic wrap, leaving some overhang to help remove the cake later.
- Press a layer of crumbled amaretti cookies into the bottom of the pan to form a crust.
Layer the Ice Cream:
- Pistachio Layer: Spread the softened pistachio ice cream evenly over the amaretti cookie crust. Sprinkle half of the chopped pistachios over the pistachio layer. Place the pan in the freezer for about 15-20 minutes to firm up.
- Cherry Layer: Spread the softened cherry ice cream over the pistachio layer. Sprinkle the chopped maraschino cherries over the cherry ice cream. Return the pan to the freezer for another 15-20 minutes.
- Chocolate Layer: Spread the softened chocolate ice cream over the cherry layer. Pour the chocolate fudge sauce over the chocolate ice cream and spread evenly. Sprinkle the remaining chopped nuts over the fudge layer. Freeze the pan for at least 2 hours or until the ice cream is firm.
Assemble the Cake:
- Once the layers are firm, remove the ice cream cake from the springform pan using the plastic wrap overhang. Peel off the plastic wrap and place the cake on a serving platter.
Finish the Cake:
- Spread whipped cream evenly over the top and sides of the ice cream cake.
- Garnish with additional maraschino cherries and sprinkles.
Serve:
- Slice the spumoni ice cream cake and serve immediately. Enjoy!
Tips:
- Ensure the ice cream is softened enough to spread easily but not melted.
- For a cleaner cut, dip your knife in hot water before slicing the cake.
- For added flavor, you can brush the amaretti cookie crust with a bit of liqueur or coffee before pressing it into the pan.
Fall 2023 Content
EM Embraces 3D Volume (vEM)
by Chad Galloway, PhD
Returning from the Microscopy & Microanalysis meeting in Minneapolis this past July, one theme was abundantly clear in the field of electron microscopy, that, 3 dimensional volume EM (vEM) which utilizes serial sectioning collection, is fast becoming a routine imaging modality, challenging 2D imaging where only single thin section views are used documenting structural changes to cells and organelles. Increasingly, these studies utilize multiple modalities, the most poignant being correlative light and electron microscopy (CLEM). These volume techniques are not novel or even newly developed, as CLEM was developed in the early 1990s. Serial sectioning EM dates back to the early 1950’s, soon after scientists first developed methods for embedding of biologic specimens for transmission electron microscopy. However, it was painstakingly slow to perform, as electron micrographs on film negatives necessitated darkroom printing on photopaper prior to performing the laborious tracing of organelles for 3-dimensional representations.
What is driving this change to vEM? Advances in technology, instrumentation, reagents, and methodologies have been continuously evolving while being complimented by the coordination of scientists in the vEM community1, democratizing this technique. Much of this is taking place in the software involved in the segmentation and reconstruction of data in an automated fashion. The vEM community defines it as newly-developed imaging using transmission or scanning electron microscopy to allow 3-dimensional investigation of cells and tissue ultrastructure up to millimeters in volume with nanometer resolution. The technique encompasses various methodologies distinguished by the nature of sectioning; Focused Ion Beam scanning electron microscopy (FIB-SEM) where the block face is shaved by a gallium or plasma beam, Serial Block Face SEM (SBF-SEM) where an ultramicrotome sections the block housed inside the SEM itself and array tomography where the individual sections are cut on an ultramicrotome and are collected serially on slides, tape and/or silicon wafers2 . In the EMR we have completed and published a study using the latter of these methodologies, interrogating the invasion of canaliculi in a S. aureus bone infection model utilizing tape collected sections on the ATUMtome and image collection in back scatter mode in an SEM3. Improvements in detection in backscatter mode and optimization of tissue preparation, to improve contrast, allow for acquisition of images almost indistinguishable, ultrastructurally, from routine transmission electron microscopy. Array tomography has the added advantage that it can be re-interrogated for multiple regions of interest (ROI). The benefit of re-interrogation is accentuated when doing CLEM. Recent developments of fixation resistant fluorescent proteins and protected probes, normally quenched by crosslinking aldehydes and osmium tetroxide, allow for fluorescent imaging post embedment, streamlining the targeting of cells/structures of interest4. A routine request of customers at the EMR is to target only those rare cells that were transfected in a mixed-cell population of a tissue, these vEM advancements will increasingly make the task of finding that “needle in a haystack” easier. As a methodology alone, vEM is becoming an essential tool in neuroscience studies whereas the routine 2D view of a 70 nm thin section, generated by ultramicrotomy, has become inadequate to describe synaptic structure and connectivity. At an intracellular level, vEM is now the preferred tool of mitochondrial researchers, where the restriction of 2-dimensional analysis can result in improper interpretation of mitochondrial shape and size descriptors. In addition to volumetric observations of mitochondria, changes in the Golgi, in a protein processing defect for example, or the ER, in the unfolded protein response, are better observed and described in 3-dimensions. These reconstructions also better describe inter-organelle contacts, critical sites for cross-talk in response to stimuli, often underappreciated in standard 2D electron microscopy.
The prospect of this electron microscopy renaissance towards 3 dimensional visualization is exciting to us in the EMR. The journal Nature agrees, naming vEM one of top 7 technologies to watch in 20235. We are familiar with the techniques and technologies and plan to move toward acquisition of the necessary instrumentation. If you have a project that would benefit from CLEM and/or vEM, we invite you to reach out for further discussion.
2 https://doi.org/10.1111/boc.201600024
3 https://doi.org/10.1002/jor.24968
4 https://doi.org/10.1016/j.cbpa.2023.102369
5https://doi.org/10.1038/d41586-023-00178-y
MSRL Interactive Data Analysis
by Kyle Swovick, PhD
INTRODUCTION
Over the past few years, the proteomics field has made huge strides in virtually ever aspect: sample prep, data acquisition, data processing, and statistical analysis. These advancements, while resulting in amazing science and uncovering new biology, it also results in ever–increasing sizes of data files. Receiving an Excel spreadsheet with protein expression levels, log2 fold changes, and p-values for nearly 10,000 proteins can be daunting for the un-initiated researcher who is not comfortable working within coding environments.
At the MSRL, as discussed in the previous newsletter, we have revamped our acquisition techniques resulting in almost a 100% increase in quantifiable proteins dramatically increasing the size of reports we were sending, resulting in increased challenges for researchers analyzing their own data. To facilitate this, we have been spending the last year building a tool that will allow researchers with no coding experience to be able to visualize and interact with their own data.
DATA DELIVERABLES
Previous Format
Long-time users of the MSRL will be well aware of the usual format of the data reports we send: Excel files with rows corresponding to:
• Protein identifiers (Gene Name, Uniprot ID, Protein Name)
• Number of peptides ID’d per sample
• Protein abundance for each sample
• If there were biological groups or conditions:
– median abundance for each group
– log2 fold change between the requested comparisons
– p-value from a student’s t-test between groups
Updated Format
From now on, for any project, MSRL users will still receive the same Excel file. Additionally, if there are any group comparisons, the user will now also receive a zipped folder that will contain an HTML document with several interactive figures (shown below) to help with understanding their data quality and differential expression. For example, by hovering over a dot in a volcano plot, you will see what that protein is along with the log2 fold change and p-value.
Included Figures
DATA QUALITY METRICS
To quickly asses the quality of the data, we use several different figures:
- A heatmap showing the correlation between samples (also clusters each sample. In an ideal world, all samples within the same group should be clustered together as shown here with the horizontal bar.
Figure 1. Correlation-based Hierarchal Clustering
2. Distributions of the CVs of protein abundances within a group (a lower CV indicates less variation within a group. Figure 2). We also include the CV when the abundances are measured across all groups. In nearly every experiment, these CVs should be higher than group-specific CVs.
Figure 2. Distribution of Protein Abundance CVs
3. Distributions of protein abundances for each sample. Ideally, these distributions should be relatively similar to each other, especially for samples within the same group.
Figure 3. Distribution of Protein Abundances
Volcano Plot
To see what proteins are differentialy expressed between different conditions, volcano plots are commonly used. These plot the log10 of the p-value against the log2 fold change in protein expression levels. In Figure 4, we have an example plot where we set a cut-off level of a log2 fold change of greater than 1 or less than -1 (equivalent to a 2x fold change in expression levels) and a p-value of 0.05, we can see a handful of proteins are either expressed higher in dKO (highlighted in dark blue) or higher in SCR (highlighted in red). Additionally, if you hover over any dot, you can see exactly what protein that refers back to.
Figure 4. Volcano Plot of dKO vs SCR.
BENEFITS TO INTERACTIVE HTML FILE
We believe that introducing these figures into our deliverable objects provide several benefits for users including:
• Reducing the time needed for researcher’s to analyze their data
• Figures researchers can include in presentations and papers (Note: in figure 4, there is a camera that you can press to make a .png of the figure.)
• In figure 1, there are two grey arrows with “Code” next to them. If those arrows are pressed, it expands to reveal the code used to generate the figures (Figure 5). This allows researcher’s who are familiar with coding (specifically R) to recreate these figures and tweak setting to better suit their own needs.
Figure 5. Expandable Code Block.
FUTURE PLANS AND UPGRADES
Making a solid foundation that we could build upon further was of great importance to us. As novel analyses, techniques, and visualization methods come along, we can implement them within this framework. In the short term for example, we are planning to include several more figures including Gene Ontology (GO) network analysis and protein family heatmaps in the near future.
FCR (Flow Core Resource) Beer Dip
by Meghann O'Brien
INGREDIENTS (yields 4 cups)
- 2 - 8 oz packages cream cheese, softened
- 3 tablespoons ranch dressing mix or 1 package ranch dressing mix
- 2 cups shredded sharp cheddar cheese
- 2 green onions, chopped
- ~ ½ cup beer
- 1 jalapeno, chopped
DIRECTIONS
- In a bowl, combine cream cheese and ranch dressing mix
- Stir in cheese, green onions, jalapeno
- Add the beer until you reach your desired consistency
- Cover and refrigerate overnight
- Serve with pretzels or crackers
"Way Too Much Work" Short-Rib Chili
By Kyle Swovick
Nothing really fends off those cold and damp WNY November days and keeps the soul warm like chili. And this chili, slightly modified from The Food Lab by J. Kenji Lopez-Alt, while a lot of work, may honestly be one of the best you’ve ever had; I’ve brought a native Texan near tears, my friends will not allow me to attend our yearly trip if I don’t bring this with me…it’s almost made a vegetarian re-think her choices.
While yes there are A LOT of ingredients and steps, I think they are all worthwhile. But if you don’t have the time before heading out to Orchard Park, or just simply don’t want to, I’ve included a few shortcuts that approximate 75% of the final product. Also, short-rib is silly expensive right now (I recommend getting some at the Asia Food Market at Brighton-Henrietta Town Line Rd) so substituting with another highly-marbled cut with lots of connective tissue like chuck is a really good option. It is also important to note that this dish gets BETTER as it sits. So it might be best to prepare the beans and meat on Friday night, cook on Saturday, and then bring the finished dish with you to the tailgate where you’ll just need to warm it up.
INGREDIENTS
• 5 lbs bone-in short rib or 3 lbs boneless short rib, trimmed of excess fat. Optionally, use 3 lbs boneless chuck.
• Salt and black pepper
• 2 tbsp vegetable or canola oil
• 1 large yellow onion, finely diced
• 1 jalapeno or 2 serrano peppers, finely chopped
• 4 cloves garlic, minced
• 1 tbsp dried oregano
• 1 cup Chile Paste (instructions below). Optionally, 1/2 cup chili powder.
– 6 nacho, pasilla, or mulato chiles, seeded and torn into 1-inch pieces
– 3 New Mexico red, California costeno, or choricero chiles, seeded and torn into rough 1-inch pieces
– 2 cascabel, arbol, or pequin chiles, seeded and torn in half
– 2 cups chicken stock
• 4 cups chicken stock (preferably homemade)
• 1 pack gelatin (if using store-bought stock, optional)
• 1/2 cup beer (preferably Labatt Blue or Genny R&W)
• 1/2 cup coffee
• 4 anchovy filets, mashed into a paste with the back of a fork
• 1 tsp Marmite (optional)
• 1 tbsp soy sauce
• 2 tbsp tomato paste
• 2 tbsp cumin seeds, toasted and ground
• 2 tsp coriander seeds, toasted and ground
• 1 tbsp unsweetened cocoa powder
• 3 tbsp instant cornmeal
• 2 bay leaves
• Kidney beans. Preferably 1lb dried, soaked in salted water at room temperature for at least 8 hrs. Optionally 2.5 lbs canned kidney beans, drained.
• 1 28 oz can crushed tomatoes
• 1/4 cup apple cider vinegar
• 1/4 cup whiskey (optional)
• 2 tbsp hot sauce
• 2 tbsp dark brown sugar
• Garnishes as desired
MAKING THE CHILI PASTE
Substituting the standard chili powder with homemade chile paste really is what brings this to a new level, so I highly recommend doing this. Not only does it improve the texture because you’re not adding a ton of chili powder, but you can fine tune the mix to include whatever chiles you want, so you can make the flavor and spice level unique to your kitchen. Also, you can just make one large batch and then put the paste into ice cube trays and then once frozen, store them in bags for a year. This way whenever you make a dish that calls for chili powder, you can sub 2 tablespoons of this paste for every 1 tablespoon of powder.
STEPS
1. Toast the chiles in a Dutch oven or stock pot over medium heat, stirring frequently, until slightly darkened, with an intense toasty aroma, 2 to 5 minutes.
2. Add the chicken stock and simmer until the chiles have softened, 5 to 8 minutes.
3. Transfer the liquid and chiles to a blender and blend, starting on low speed and gradually increase the speed to high, scraping down the sides as necessary, until a completely smooth puree is formed, about 2 minutes. Add water if the mixture is too thick to blend. Let cool.
MAKING THE CHILI
The Night Before Cooking (completely optional):
• If using dried kidney beans: Add the beans to enough salted room temperature water to cover by several inches (the beans will soak up the water and expand overnight).
• Pat the beef dry and season all over with plenty of salt (think about how a sidewalk looks after it’s been snowing for 15 minutes). Place on a wire rack over a baking sheet in the fridge and let sit.
Cooking Day
1. Season the beef on all sides with pepper and salt (if not salted overnight). Heat the oil in a large Dutch oven over medium-high heat until smoking. Add half of the meat and brown well on all sides (depending on the size of your pot, you may need to do more than 2 batches. It is important to not crowd the pot to ensure good browning), reduce the heat if the fat begins to smoke excessively or the meat begins to burn.
2. Transfer to a plate and repeat step 1 with the remaining meat.
3. Reduce the heat to medium-low, add the onion, and cook, scraping up the browned bits from the bottom of the pan with a wooden spoon and then stirring frequently, until softened but not browned (6-8 minutes).
4. Optional: if using store-bought stock, pour into a dish and pour a packet of gelatin over top and let it bloom. (This only improves the mouth-feel of the final dish and will not impact the flavor so feel free to skip)
5. Add the fresh chile, garlic, and oregano and cook, stirring, until fragrant (~ 1 minute).
6. Add the chile paste and cook, stirring and scrapping constantly until it leaves a coating on the bottom of the pot (2-4 minutes).
7. Add the chicken stock and scrape any browned bits from the bottom of the pot.
8. Add the anchovies, beer, coffee, soy sauce, tomato paste, ground spices, cornmeal, and Marmite if using. Whisk to combine and keep warm over low heat.
9. Adjust an oven rack to the lower-middle position and preheat the oven to 225F.
10. Remove the meat from the bones and reserve the bones (if using bone-in short-rib). Chop all the meat into rough 1/4-1/2 inch pieces.
11. Add any accumulated juices from the cutting board to the dutch oven and add the chopped beef (and bones if you have them), and bay leaves to the chili.
12. Bring to a simmer, cover, and place in the oven for 1 hr.
13. If using dried beans: Drain the beans and transfer to a pot and cover with water by 1 inch. Season with salt and bring to a boil over high heat then reduce to a simmer and cook until the beans are nearly tender (about 45 minutes). Drain.
14. Remove the chili from the oven and add the tomatoes, vinegar, and beans.
15. Return to the oven with the lid slightly ajar and cook until the bean and beef are tender and the stock is rich and slightly thickened (1.5-2 hrs longer). Add water if necessary to keep the beans and meat mostly submerged (a little bit poking out is OK).
16. Remove the bay leaves and bones. Add the whiskey, hot sauce, and brown sugar and stir to combine. Season to taste with salt, pepper, and vinegar.
17. Let sit overnight in an airtight container.
Game Day
• Pat the beef dry and season all over with plenty of salt (think about how a sidewalk looks after it’s been snowing for 15 minutes). Place on a wire rack over a baking sheet in the fridge and let sit.
Spring 2023 Content
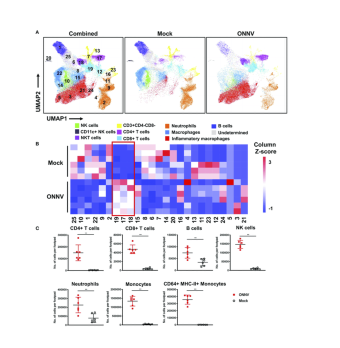
Our Flow Cytometry Resource Offers High-Dimensional Analysis
by Jim Java
Flow cytometry experiments often conclude with the production of FCS computer files containing investigators' raw results. CART's Flow Cytometry Resource (FCR) can now provide standardized, reproducible analyses of FCS files from flow experiments.
After a flow cytometry experiment, it's not uncommon for investigators to import their FCS files into software such as FlowJo or FCS Express, and then proceed with an analysis "by hand", which can be time-consuming and somewhat subjective. In the interest of saving time and limiting subjectiveness, the FCR data analysis team has developed a soup-to-nuts analysis pipeline for the R programming environment: we call it "flowpipe" and it can semi-automatically handle most analytical tasks from pre-processing/pre-gating to phenotype clustering to differential-expression modeling.
The results of a flowpipe run include UMAP visualizations, spreadsheets summarizing the phenotype clusters, per-cluster FCS files, and a detailed summary report of sample or group differences. Although we developed flowpipe to be useable by researchers savvy of R programming (and we're glad to help you set it up!), we recommend that you request a flowpipe run as part of your FCR scheduling, so that our data-analysis team can manage the process.
The length of a flowpipe analysis depends on the number and size of the FCS files provided to the software, but a typical run takes a few hours. Our software has aggregated a number of common techniques and algorithms (well-represented in the peer-reviewed literature) into a flexible parallel-processing framework that's meant to reduce investigators' analytical workload; so, contact us if you'd like more information about sending your FCS files through the flowpipe pipeline.
As part of the flowpipe analysis process, we ask investigators to provide "metadata" relevant to their flow-cytometry experiment: that is, for example, whether samples are cases or controls; a list of pre-defined phenotype gates for drilling down to interesting cell subsets; and patient data that can be incorporated into the differential-expression models. For more information, please check out the flowpipe GitHub respository or contact Jim Java. We can also provide statistical analyses outside the scope of the pipeline—inquiries are welcome!
Our Mass Spectrometry Resource Laboratory Overhauls Proteomic Data Acquisition
by Kyle Swovick, PhD
Over the past two years, the MSRL has been undergoing an overhaul of our proteomic data acquisition methods.
For decades, proteomic data has been primarily collected through data-dependent acquisition (DDA). In this method, the mass spectrometer isolates and fragments just a single peptide for identification and then repeats this process throughout the entire gradient. Recently, a method termed data independent acquisition (DIA) has been introduced that promises vast improvements over DDA proteomics.
When performing DIA experiments, the mass spectrometer isolates and fragments every peptide within a predefined mass range. Performing fragmentation and identification this way, in theory, offers several benefits including greater coverage and less missing values. These gains are primarily a result of the stochastic nature of peptide fragmentation in DIA. DDA proteomic experiments historically have been plagued by high intensity peptides since those will have a greater propensity to be chosen for fragmentation, thereby ignoring many lower intensity peptides.
DIA experiments however alleviate this problem; regardless of a peptide’s intensity it will be fragmented, thus leading to more possible fragment ions that can be used for identification. When the instrumentation improvements offered by DIA are paired with the recent advancements of neural network and machine learning programming, the results are truly extraordinary.
Using cutting-edge techniques, the MSRL saw nearly 100% and 50% increase in tissue and cell culture samples respectively (Figure 1A). Their DIA pipeline also saw a 25% improvement in data completeness meaning (Figure 1B).
Combined, these improvements mean users can result in up to a 150% improvement in their calculations when measuring differentially expressed proteins. If you are curious about what kind of coverage DIA can yield your specific biological matrix, Table 1 includes many of the common sample types the MSRL handles. And, if you are intrigued by what DIA can offer for your own research, you can reach out to MSRL with any questions.
Stay tuned for the next installment where the MSRL will talk about the improvements they’ve made to their data analysis pipeline to help researchers delve into and interact with their data.
Experiments in the Kitchen: Beer in the Sheath Tank
by Steven Polter
Whether or not some of us want to admit it, we all still play pretend in some way or other. I, myself, enjoy to pretend I am a brewmaster. I’ve been a homebrewer for years and many of my associates, including my brewing partner, will tell you that my relationship with brewing flirts with the line between hobby and habit. In 2022 I had the pleasure of being asked to play brewmaster for CART and provide a few kegs of beer for a retreat last July. Recently I have been asked to don the mask once again to share and discuss one of those recipes, so I chose to share Supercrisp 570, an American Kolsch.
Perfect for the spring days just ahead of us, this beer beckons the reawakening from winter’s dim but cozy torpor. Bright straw yellow and exuberant, Supercrisp 570 positively pops with a floral nose and lemon-citrus flavor backed by pleasant, bready malt. This brew was designed to shine no matter when or where you drink it!
Ingredients (For a 5-gallon batch)
- 12 lb. 2-Row Brewer’s Malt (milled)
- 3 oz. Lemondrop Hops (T-95 pellets, 2 oz. for the boil and 1 oz. for dry hop)
- 1 pouch WLP 810 San Francisco Lager Yeast
https://www.whitelabs.com/yeast-single?id=220&type=YEAST&style_type=2 - Water (~7.5 gal. total)
Brewing notes
- Set yeast and hops aside to come to room temperature during the process
- Step mash* with 4 gal. H2O. USE LOW HEAT AND STIR CONSTANTLY WHEN RASING THE MASH TEMPERATURE TO THE NEXT STEP! THERE WILL BE NOSCORCHING!
- Heat H2O to 135F* and add milled malt. Stir to mix well. Rest 20 minutes at 125 F
- Raise temp and rest 30 minutes at 140 F
- Raise temp and rest 30 minutes at 150 F
- Mash out and set sweet wort aside
- Sparge with 3.5 gal. H2O at 170 F for 10 minutes. Recirculate/vorlauf until the wort runs clear after the 10 minute rest, then sparge out into your kettle containing the sweet wort from the first run
- Boil 60 minutes. Add hops as follows:
- 1 oz Lemondrop 60 minutes (this notation means the hops spend the listed amount of time in the boiling beer, in this case these hops are added just after the wort begins to boil)
- 1 oz Lemondrop 30 minutes
- When boil is complete, cool wort to ~70 F
- With clean hands and using a clean, sanitized funnel, transfer the wort to a clean and sanitized fermentation vessel. Take a sample of wort at this point for testing of specific gravity. Place a foil cap over the mouth of the vessel after the wort is transferred while the yeast is readied for pitching
- Again with clean hands and using clean/sanitized scissors, cut the yeast pouch carefully over the open mouth of the fermentation vessel and gently, carefully pitch the yeast into the wort
- Replace the foil cap over the mouth of the vessel and CAREFULLY shake the vessel vigorously for 30 seconds to 1 minute (this serves to oxygenate the wort which is crucial for initial yeast health/activity as well as mix things up nicely)
- Ready a clean, sanitized airlock and stopper assembly and quickly peel back the foil cap and place the stopper/airlock combo firmly into the mouth of the vessel
- Label your vessel (I speak from experience) with the name, date, and original gravity of the wort
- Give your vessel a gentle slap on the side and take a moment to feel accomplished, maybe crack a beer
- Consider covering your fermentation vessel with an old t-shirt or whatever else will help keep light out of it. Seriously, being a fungus yeast is not in love with bright light or direct sunlight. Definitely not bright, direct sunlight, which will also zap your tasty, hard-earned flavor compounds!
* A step mash is a technique that entails resting the mash at increasing temperature steps to maximize sugar extraction and provide a greater breadth of sugar types in the wort as well as leaving some non-fermentable sugar, which provides a pleasant, bready sweetness in the finished beer. When heating the water for the first step in the mash, be sure to overshoot by about 10 degrees F, as the thermal mass of the grains when added to the water (in these relative volumes) will sink about 10 degrees off the temperature of the mash after mixing.
Fermentation
- Ferment at ~60 F (basement/cellar temperature is prime for this!) for 3 weeks
- Transfer to secondary fermentation and add 1 oz Lemondrop. Secondary for 19 to 21 days, still at ~60 F
- Transfer to keg and pressurize to begin carbonation. If possible, place the keg into a temperature controlled lagering chamber and step the temperature down by 2-3 degrees F each day until it reaches 35-36 F. During this process, pressurize the keg each day (to somewhere around 25 PSI) to slowly carbonate the beer while you cold-condition it. If there is no access to a temperature controlled lagering chamber the keg can be pressurized and cold-conditioned in a regular old refrigerator or kegerator without temperature control. The beer will still be good!
- Cold-condition and carbonate in this way until desired carbonation level is reached. Cold-conditioning can be continued after carbonation level is reached, up to 3-4 weeks. Periodically draw small amounts of beer from the keg to pull out any sediment that has crashed to the bottom of the keg, and to taste, of course!
- Serve and enjoy!
If you are not experienced in brewing and have question marks for any reason, feel free to reach out to me at Steven Polter and I will happily discuss, clarify, and provide additional information!