Channelopathies and Skeletal Dysplasia
Elucidation of mutated TRPV4 ion channel differentially regulate complex gene regulatory networks governing cartilage and bone development, as well as how mutated TRPV4 alters tissue homeostasis in response to mechanical loading.
TRPV4, a polymodal Ca2+-permeable ion channel, functions as a primary mechanosensor in chondrocytes (Fig 1A). Mutations in TRPV4 can lead to abnormal cartilage/skeletal formation in humans; however, the mechanisms by which TRPV4 mutations cause these pathogenic changes in cartilage at the cellular and molecular levels is largely unknown. The overall goals of this project are to utilize several innovative techniques including gene editing, Next-generation sequencing, and bioinformatic analyses to unravel mechanisms by which TRPV4 regulating chondrocyte maturation and cartilage homeostasis in response to mechanical loading, with particular focus on how distinct TRPV4 mutations disrupt these cellular pathways.
Recently, using CRISPR-Cas9 genome editing, we have created isogenic human induced pluripotent stem cell (hiPSC) lines harboring distinct mutations in TRPV4 that lead to varying degrees of skeletal dysplasia, including V620I (mild severity) and T89I (neonatally lethal) (Fig 1B). These hiPSCs lines can then be differentiated into chondrocytes using our previously established protocols (Fig 1C). Significantly increased Ca2+ signaling was observed in the chondrocytes bearing T89I mutation (Video 1-3). By applying single-cell RNA sequencing (scRNA-seq) and the TRPV4-mutated hiPSC lines, we will be able to elucidate how TRPV4 mutations dysregulate transcriptome profiles and gene regulatory network governing chondrocyte maturation. We will further determine how TRPV4 mutation alters the biologic response of chondrocytes to dynamic mechanical loading and identify selective TRPV4 antagonists that can restore such alternations (Fig 1D). Currently, no therapies are available for TRPV4 mutation-associated lethality or skeletal malformations. Thus, the results of this work will provide novel insights into the development of new pharmacologic therapies for TRPV4-associated channelopathies.
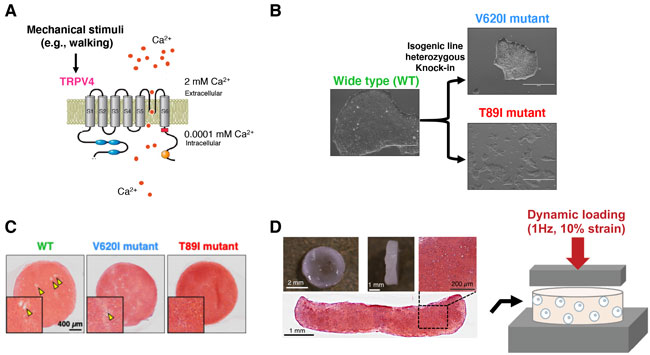
Fig 1. (A) Schematic model of TRPV4 ion channel. (B) Isogenic hiPSC lines harboring distinct mutations in TRPV4 that lead to varying degrees of skeletal dysplasia, including V620I (mild severity) and T89I (neonatally lethal) generated via CRISPR-Cas9 genome editing. (C) hiPSC-derived chondrocytes withT89I mutant demonstrated decreased chondrocyte hypertrophy compared to wide type (WT) chondrocytes. (D) Dynamic mechanical loading model and scRNA-seq to elucidate how TRPV4 mutation alters the biologic response of chondrocytes to loading and identify selective TRPV4 antagonists that can restore such alternations.
Videos
Wide type human chondrocytes
TRPV4 (V620I)-mutated human chondrocytes
TRPV4 (T89I)-mutated human chondrocytes
Video Legend: hiPSC-derived chondrocytes were treated with GSK101 (specific TRPV4 ion channel agonist) to activate Ca2+ signaling. Once the dye (Fluo-4) in the cell detects the Ca2+ influx, the cell lights up in green color.