




Welcome to the Land Lab
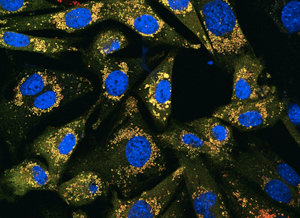
Cancer cells: blue is cell nucleus
and yellow represents the garbage recycling process
that occurs as cancer grows.
Cooperation of oncogenic mutations in the control of malignancy:
- Understand the molecular basis for oncogene cooperation
- Identify unique features of gene networks in cancer cells vs. normal cells
- Exploit such features of cancer cells for targeted approaches towards therapeutic intervention
- Systems biology of cancer cells
Carcinogenesis is a process driven by multiple co-operating oncogenic mutations in which features of the cancer cell phenotype, such as proliferative advantage, only emerge as a result of the interplay between these mutations. This is consistent with the notion that many divergent cancers share a limited number of disease mechanisms with common underlying signaling networks, despite the many genes implicated in the disease.
The Land laboratory has pioneered investigation of the nature and underlying principles of cancer gene cooperation originating with the discovery that multiple oncogenic mutations are required for malignant cell transformation. Using a wide variety of experimental approaches, including genomics and systems biology, the laboratory has demonstrated that the cooperation of cancer-promoting genetic lesions is strongly reflected by synergistic modulation in signaling and the gene networks of malignant cells.

Hartmut K. Land, Ph.D.
Principal Investigator
Selected Publications
Smith, B., Schafer, X. L., Ambeskovic, A., Spencer, C. M., Land, H. and Munger, J. (2016) Addiction to Coupling of the Warburg Effect with Glutamine Catabolism in Cancer Cells. Cell Reports 17: 821-836. PMCID: PMC5108179
Komisarof, J., McCall, M., Newman, L., Bshara, W., Mohler, J.L., Morrison, C. and Land, H. (2017) A Four Gene Signature Predictive of Recurrent Prostate Cancer. Oncotarget, 8: 3430-3440.
Kinsey, C., Balakrishnan, V., O’Dell, M. R., Huang, J. L., Newman, L., Whitney-Miller, C. L., Hezel, A. F. and Land, H.. (2014). Plac8 Links Oncogenic Mutations to Regulation of Autophagy and Is Critical to Pancreatic Cancer Progression. Cell Reports, pii: S2211-1247(14)00258-7. doi: 10.1016/j.celrep.2014.03.061. [Epub ahead of print]
Sampson E.R., McMurray, H.R., Hassane, D.C., Newman, L., Salzman, P., Jordan C.T. and Land, H.. (2013). Gene signature critical to cancer phenotype as a paradigm for anti-cancer drug discovery. Oncogene, 32: 3809-3818. Epub 2012 Sep 10.
Ashton, J.M., Balys, M., Neering, S.J., Hassane, D.C., Cowley, G., Root, D.E., Miller, P.G., Ebert, B.L., McMurray, H.R., Land, H. and Jordan. C.T. (2012). Gene sets identified with oncogene cooperativity analysis regulate in vivo growth and survival of leukemia stem cells. Cell Stem Cell 11: 359-372.
Smith, B and Land, H. (2012). Tumor suppressor activity of the cholesterol exporter gene Abca1. Cell Reports 2: 580-590.
McMurray, H.R., Sampson, E. R., Compitello, G., Kinsey, C., Newman, L., Smith, B., Chen, S., Klebanov, L., Salzman, P, Yakovlev, A and Land, H. (2008). Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature, 453:1112-1116.
Xia, M. and Land, H. (2007). Tumor suppressor p53 restricts Ras stimulation of RhoA and cancer cell motility. Nature Structural & Molecular Biology 14:215-223.
More papers:
Lab Members






News
Affiliations
- Biomedical Genetics
- Biochemistry & Biophysics
- Center for Biomedical Informatics
- James P. Wilmot Cancer Center
- UR Stem Cell and Regenerative Medicine Institute
- Biochemistry & Molecular Biology Ph.D. Program
- Biomedical Data Science
- Biomedical Genetics and Genomics Ph.D. Program
- Translational Biomedical Science Ph.D. Program
- Zhao Lab
June 18, 2019
A Graphic Design Revolution For Scientific Conference Posters
August 2, 2018
Hucky Land Leads Genome Sequencing Project that Expands Tissue-Banking Partnership
December 20, 2016
Research Led by Hucky Land Points to Prostate Cancer Tool
November 15, 2016
Wilmot Co-directors Honored with Davey Award
Contact Us
Land Lab
601 Elmwood Ave
Rochester, NY 14642
(585) 273-1450