URMC / Labs / Phizicky Lab / Projects / The Biological Roles of tRNA Modifications
The Biological Roles of tRNA Modifications
Investigation of the biology of Trm7 and 2'-O-methylation of the anticodon loop
We are interested in the biology of Trm7 because of its widespread importance in eukaryotes, and because its important function is not yet known. Deletion of TRM7 in each of the evolutionarily distant fungi S. cerevisiae and Schizosaccharomyces pombe results in a severe growth defect, and humans with lesions in the orthologous gene FTSJ1 have non-syndromic X-linked intellectual disability. In S. cerevisiae,Trm7 interacts with its partner proteins Trm732 and Trm734 to catalyze 2’-O-methylation of residues N32 and N34 in the anticodon loop of its three substrate tRNA species, and the resulting Nm32 and Nm34 modifications in one of its substrates (tRNAPhe) promote formation of wybutosine from m1G37 in the anticodon loop. Moreover, tRNAPhe appears to be the biologically important substrate in S. cerevisiae, since overproduction of this species overcomes the growth defect of a trm7Δ mutant. This architecture for tRNAPhe anticodon loop modification, involving Trm7 interactions with Trm732 and Trm734, and the Nm32, Nm34-driven wybutosine formation in tRNAPhe, is conserved in S. pombe and humans, and tRNAPhe is the biologically important substrate in S. pombe. Thus, we speculate that tRNAPhe may be the biologically important human FTSJ1 substrate that leads to non-syndromic X-linked intellectual disability.
Our ongoing research on Trm7 is focused on the understanding its biological roles. To understand its roles in S. cerevisiae, we isolated spontaneous suppressors of the slow growth defect of trm7Δ mutants. We found that most mutations mapped to subunits of phenylalanyl tRNA synthetase, which catalyzes charging of tRNAPhe to form phe-tRNAPhe. This result was surprising as there was little detectable charging defect in trm7Δ mutants for any of the three Trm7 tRNA substrates, as measured using a standard biochemical assay. Nonetheless, we found evidence for robust constitutive activation of the general amino acid control (GAAC) pathway in trm7Δ mutants, which senses uncharged tRNA to phosphorylate eIF2α and alter expression of a large number of genes. Moreover, mutations in PheRS that suppressed the trm7Δ slow growth phenotype concomitantly reduced activation of the GAAC response, and treatments predicted to channel tRNAPhe toward translation all suppressed the growth defect and reduced activation of the GAAC response. Consistent with a conserved biological effect in eukaryotes, our genetic analysis in S. pombe yielded almost identical results: There was no detectable charging defect in trm7Δ mutants by standard assay, but there was robust activation of the GAAC pathway, and suppressors of the growth defect mapped to subunits PheRS, and reduced GAAC activation.
Our current research on Trm7 biology is focused on understanding how the GAAC response is constitutively activated in S. cerevisiae and S. pombe trm7Δ mutants in the absence of any obvious charging defect, and on understanding the properties of PheRS variants that improve growth of trm7Δ mutants
Investigation of the rapid tRNA decay (RTD) quality control pathway
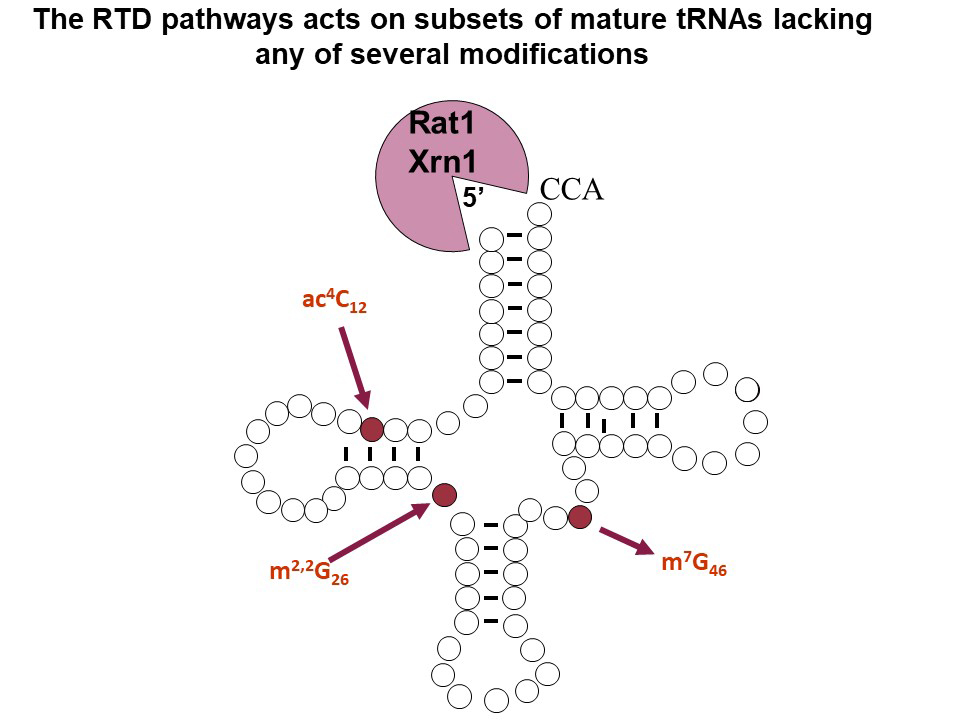
We are interested in tRNA decay mechanisms because several ubiquitous body modifications have been implicated in maintaining the stability of tRNA in vivo and because maintaining sufficient levels of each tRNA species is crucially important for normal cell growth. Two quality control pathways described in S. cerevisiae are known to target decay of tRNA due to lack of modifications, to the presence of destabilizing mutations, or to defects in folding. The nuclear surveillance pathway targets both pre-tRNAiMet lacking the m1A58 modification and a significant portion of the total transcribed pre-tRNA, acting through the TRAMP complex and the nuclear exosome to degrade pre-tRNAs from the 3' end. The rapid tRNA decay (RTD) pathway, discovered earlier in this lab, targets a subset of mature tRNA species lacking any of several body modifications, and mature tRNAs with destabilizing mutations, all of which expose the 5' end for decay by the 5'-3' exonucleases Rat1 and Xrn1. RTD is inhibited by elevated levels of the metabolite adenosine 3',5' bis-phosphate that are found in met22Δ mutants, and is in competition with translation factors that can sequester the tRNA from RTD.
Although activation of RTD by 5' end exposure is well established, it is seemingly at odds with the results of our high throughput analysis of RTD among variants of a nonsense suppressor tRNA. This analysis showed unexpectedly that mutations in the anticodon stem-loop could provoke decay by the RTD pathway, which was surprising as mutations in this region were not expected to significantly interact with the tRNA fold to expose the 5' end to the RTD pathway.
Current work is focused on understanding how this prominent class of anticodon stem-loop variants provokes decay by the RTD pathway. In addition, we are exploring the extent to which RTD and RTD mechanisms are conserved in other organisms.
« back to all projects