Glial Responses to Traumatic Brain Injury, Stroke, the Role of Gliosis
Frank insult to the central nervous system (CNS), in the form of traumatic brain injury (TBI) or cerebral infarction (stroke), exposes the brain and spinal cord’s distinct cellular populations to an environmental catastrophe. Cell membranes and blood vessels rupture, spilling cytotoxic and inflammatory agents into the extracellular space and disrupting the blood supply, while blood brain barrier breakdown leads the influx of unfiltered blood born constituents, shutting down regular cellular metabolism. Such injuries result in highly variable functional outcomes depending on their location and severity and often result in intractable physical and emotional suffering, cognitive decline in the case of brain injuries, and the onset of dementia. Remarkably, despite the fact that most neuronal cell populations do not regenerate, trauma to the CNS is often followed by restoration of disrupted sensory and motor function. But on a cellular level, how does the body’s most sensitive tissue respond to trauma and what factors facilitate post-injury neuroplasticity?
Ex vivo image of diffuse microinfarction induced by carotid artery infusion of cholesterol crystals.
GFAP (Blue) indicates reactive astrocytes, CD68 (Red) indicates activated microglia.
Key issues in translational neuroscience are a lack of understanding in the role that glial cells play in normal physiology and by extension the consequences of astrocytic dysfunction in pathophysiological conditions. Because complex cognitive and executive functions heavily depend on the brain’s interconnected neural network, localized injuries to one region often produce profound global effects on function. Our interest in the role of astrocytes following CNS injuries stems from the fact that they constitute therapeutic targets that occupy prime real-estate: by extending processes that interface with blood vessels and neuronal synapses, as well as forming physically coupled networks with other astrocytes, they effectively couple nutritional support with information transfer and processing. Moreover, emerging evidence suggests that the role of astrocytes is more active and dynamic than previously considered. Astrocytes have been shown to exert control over directing cerebral blood flow (hyperemia), mediate the clearance of pathologically relevant proteins (glymphatic system), and engage in sophisticated neuro-glial signaling interactions.
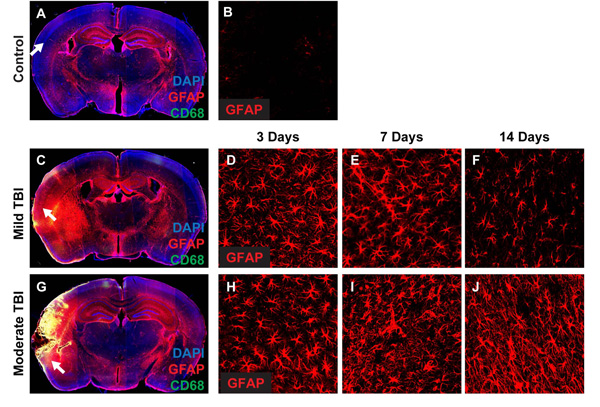
Mild vs. moderate rodent model of Hit and Run closed skull TBI, showing the time course of reactive gliosis.
Reactive astrocytes are stained for GFAP (red), while activated microglia are stained for CD68 (green).
(Image and concept for Hit and Run TBI provided courtesy of Dr. Zeguang Ren, MD. PhD.).
Despite being highly prevalent the pathogenesis of CNS injuries remains poorly understood. Experiments in our lab and others have suggested regardless of the source, acute CNS injuries evoke a similar cellular response: astrocytes and other glial cells such as microglia become activated in a process known as reactive gliosis. Although this phenomenon— first described almost a hundred years ago— is ubiquitous to a number of neuropathologies and has been extensively studied, the exact function of gliosis (whether it’s pathological or protective), its mechanisms of initiation, and its temporal relevance to therapeutic interventions following CSN injury remain controversial.
Work in our lab along with similar findings from independent labs have shown that in response to tissue trauma or non-physiological changes in the extracellular environment, astrocytes enlarge, proliferate in number, release long range signaling molecules such as ATP, and upregulate the expression intermediate filament proteins (e.g. GFAP) that are important for modifying cellular structure and astrocytic networks. Further complicating the picture of gliosis are results from experiments on animal models of traumatic brain injury, spinal cord injury and stroke, which have reported conflicting results as to its nature. These studies suggest that activation of astrocytes represents both a protective response aimed at ameliorating and stopping the spread of damage from the primary injury, as well as a pathological process that initiates secondary injury and profound disruptions to healthy function.
AQP4 de-polarization in reactive astrocytes
after Hit and Run closed skull TBI.
Although the exact role of gliosis is not yet known, the development of animal models of injury and disease have aided tremendously in characterizing and interpreting the pathological significance of glial responses to CNS injuries. Routinely, however, successful treatments performed on animals fail to predict human outcomes. One of the major challenges is a lack of models that accurately reproduce relevant clinical features of CNS pathologies. For example, many animal models of TBI—the leading cause of injury related death in the United States—rely on fixation of the head and opening of the skull to facilitate direct cortical insult, as well as large doses of anesthesia. By and large, these injuries fail to reproduce one of the least understood components of common TBI pathologies, i.e. contracoup injuries, which result in damage both to the hemisphere of impact and the contralateral side.
In response to these shortcomings, we developed a novel Hit and Run model of closed skull traumatic brain injury that reproduces the clinical features of TBI, including contracoup injuries. Interestingly, we found that moderate and severe contusions to the intact rodent skull resulted in widespread astrogliosis (on both the ipsi- and contra- lateral hemispheres) and a significant disturbance in the typically polarized expression pattern of the astrocytic water channel aquaporin-4 (AQP4).
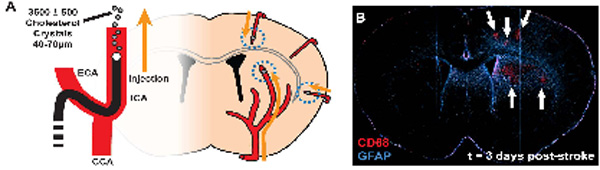
Schematic and 3 day representative image of diffuse lacunar infarction model in rodents.
Reactive astrocytes are indicated by GFAP (blue), activated microglia by CD68 (red).
Similarly lacking are models for diffuse stroke, a pathology that is marked by cognitive decline and is thought to affect as large as 30% of the broader aged population. To gain a better understanding, we developed a model where we infused cholesterol crystals to induce multiple mini-strokes (diffuse lacunar infarctions) in rodents. In a surprising finding, we noted that unlike a typical macro-infarct where neuronal death in the affected tissue is largely complete within hours, micro-infarcts triggered delayed neuronal death that progressed gradually from ˜35% at 3 days to ˜60% at 28 days. Similar to our findings from ‘Hit and Run’ TBI, this time course coincided with widespread astrogliosis and corresponding global disruptions in AQP4 polarization. Such findings suggest that injury-specific responses of astrocytes may be the key to understanding how a localized tissue insult can lead to chronic cell death and global dysfunction.
Consequently, it is not surprising that astrocytes play important roles in the pathogenesis of various disease states including the primary and secondary stages of traumatic injury or cerebral infarct. Current work focuses on identifying therapeutic windows for altering the degree of injury-induced astrogliosis, as well as evaluating the functional consequences of the breakdown of pathologically relevant solute clearance via the glymphatic pathway.
Further Reading
'Hit & Run' model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, Giese RN, Wang B, Shi X, Nedergaard M. J Cereb Blood Flow Metab. 2013 Jun;33(6):834-45.
Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. Wang M, Iliff JJ, Liao Y, Chen MJ, Shinseki MS, Venkataraman A, Cheung J, Wang W, Nedergaard M. J Neurosci. 2012 Dec 12;32(50):17948-60.
Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Chen MJ, Kress B, Han X, Moll K, Peng W, Ji RR, Nedergaard M. Glia. 2012 Nov;60(11):1660-70.
Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. Huang C, Han X, Li X, Lam E, Peng W, Lou N, Torres A, Yang M, Garre JM, Tian GF, Bennett MV, Nedergaard M, Takano T. J Neurosci. 2012 Mar 7;32(10):3333-8.
Pericyte constriction after stroke: the jury is still out. Vates GE, Takano T, Zlokovic B, Nedergaard M. Nat Med. 2010 Sep;16(9):959.
A central role of connexin 43 in hypoxic preconditioning. Lin JH, Lou N, Kang N, Takano T, Hu F, Han X, Xu Q, Lovatt D, Torres A, Willecke K, Yang J, Kang J, Nedergaard M. J Neurosci. 2008 Jan 16;28(3):681-95.
Cortical spreading depression causes and coincides with tissue hypoxia. Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M. Nat Neurosci. 2007 Jun;10(6):754-62.
Angiogenic inhibition reduces germinal matrix hemorrhage. Ballabh P, Xu H, Hu F, Braun A, Smith K, Rivera A, Lou N, Ungvari Z, Goldman SA, Csiszar A, Nedergaard M. Nat Med. 2007 Apr;13(4):477-85.
This project is supported by funding from NIH – National Institutes of Neurological Disorders and Stroke. (Failure of Metabolite Clearance in a Model of Multi-Lacunar Infarcts).
Project Collaborators:
Dr. Anthony Petraglia, Dr. Zeguang Ren