Our Research

The Serra-Moreno lab studies the mechanisms by which human pathogens such as Human Immunodeficiency Virus (HIV), its close relative Simian Immunodeficiency Virus (SIV), and SARS-CoV-2 (the causative agent of COVID-19) circumvent the barriers of the innate immunity in their respective hosts and cause disease. Current projects in the lab are investigating the interplay between these human pathogens and the cellular factors Tetherin/BST2, SERINC5, BCA2 and autophagy.
Projects on HIV/SIV
Tetherin is an integral membrane protein that traps nascent enveloped virions to the plasma membrane, thereby, impeding their release and spread. Our previous studies demonstrated that while pandemic HIV-1 use Vpu and HIV-2 use Env, most SIVs use the Nef accessory protein to overcome restriction by non-human primate Tetherin. Nef achieves this by targeting a five amino-acid motif that is present in simian Tetherin but not in the human ortholog of this protein, which may explain why HIV-1 and HIV-2 needed to evolve alternative mechanisms (Vpu, Env) for their adaptation to human Tetherin (Jia and Serra-Moreno et al., PLoS Pathog. 2009; Serra-Moreno et al., Cell Host & Microbe 2011; Serra-Moreno et al., PloS Pathog. 2013). These observations highlight the extraordinary plasticity of the primate lentiviruses to counteract this restriction factor and suggest that Tetherin constitutes a significant obstacle for infectivity and cross-species transmission.
BCA2 (Breast-Cancer Associated gene 2) is a co-factor in the restriction imposed by Tetherin on HIV. By interacting with Tetherin, BCA2 promotes the internalization and degradation of “tethered” virions. However, we found that BCA2 also exhibits Tetherin-independent antiviral activity. Specifically, BCA2 promotes the lysosomal degradation of the HIV protein Gag. Gag is the major driver of virion assembly and release. Therefore, the BCA2-mediated depletion of Gag significantly reduces virion production (Nityanadam and Serra-Moreno, PLoS Pathog. 2014). In addition to this effect, we recently uncovered that BCA2 is intimately connected with the innate immune system, since it modulates the transcription factor NF-κB. NF-κB is normally activated in response to infections and other insults. However, HIV takes advantage of the activation of this pathway, and uses NF-κB, to enhance the expression of its genes. BCA2, on the other hand, potently shuts down NF-κB signaling, significantly impairing virus gene expression and replication (Colomer-Lluch and Serra-Moreno, J. Virol. 2017). Therefore, these observations provide additional mechanisms by which BCA2 represents a promising factor for antiretroviral therapy against HIV. In fact, we are currently investigating the potential of encapsulating BCA2 as a strategy to reduce active HIV replication and enforce a permanent state of transcriptional inactivity (Fig. 1). These studies are currently funded by a NIH/NIAID R21.

Figure 1
Autophagy is a highly conserved cellular response against stress such as viral infections. The role of autophagy in HIV infection has remained controversial in the last decade. Some reports state that HIV activates autophagy to enhance its infectivity, while others claim that HIV blocks specific steps in the autophagy machinery to prevent viral degradation. We recently found that autophagy poses a hurdle for HIV by targeting Gag for elimination (Fig. 2; top). However, the virus uses Nef to circumvent this barrier. In particular, Nef intersects with autophagy at the initiation and maturation steps by blocking BECN1, although through two independent mechanisms. We recently uncovered that Nef enhances the association between the autophagy initiator BECN1 and its natural inhibitor BCL2, consequently impairing autophagosome biogenesis and restoring Gag levels (Fig. 2; bottom). Nef achieves this by recruiting the cellular E3 ligase Parkin and facilitating the mono-ubiquitination of BCL2, a post-translational modification that renders BCL2 more stable and enhances in turn its inhibitory effect over BECN1. Phylogenetic analyses of closely related lentiviral species revealed that Nef’s ability to inhibit autophagy initiation is primarily observed among the most widely distributed pandemic clades of HIV-1 and their direct ancestors SIVcpz, which suggests that autophagy antagonism might have facilitated the spread of pandemic HIV-1 viruses in the human population (Castro-Gonzalez et al., Autophagy, 2020). We are currently investigating the role of autophagy antagonism in HIV pathogenesis as well as the structural and genetic determinants that dictate Nef’s actions over autophagy initiation versus maturation. These studies have recently been funded by an HARC (HIV accessory and regulatory complexes) Collaborative Fund award.
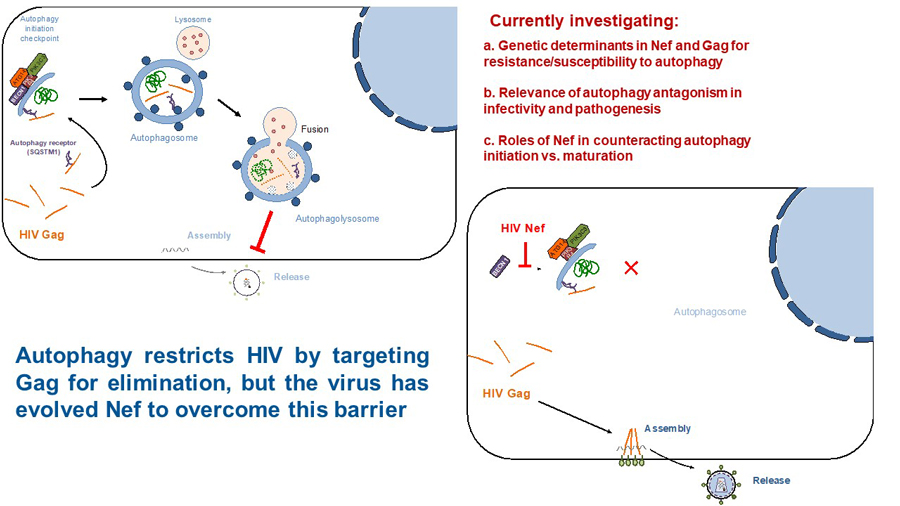
Figure 2
Projects on SARS-CoV-2
The health emergency caused by the COVID-19 pandemic urges immediate actions to elucidate the mechanisms that have contributed to the spillover and dissemination of SARS-CoV-2, its causative agent. Similar to SARS-CoV, SARS-CoV-2 emerged after the transmission of a betacoronavirus naturally infecting bats to an intermediate host (i.e. civets, ferrets, pangolins), and from the intermediate host to humans. Most viral cross-species transmissions result in dead ends, since the leaked virus is not adapted to the new host. However, the fact that coronaviruses, including SARS-CoV, MERS-CoV and SARS-CoV-2, have successfully crossed the species barrier suggests that betacoronaviruses find the necessary elements to ensure their propagation in the natural, intermediate and human host. Autophagy is a bona fide cellular process that coronaviruses usurp for their replication, and this machinery is highly conserved across mammals. Coronaviruses use autophagy as a source to generate viroplasms: double membrane structures where virus genome replication takes place. In addition to viroplasms, coronaviruses also remodel cellular membranes to create double membrane vesicles (DMVs) to shield intermediate products of genome replication –including double-stranded RNA (dsRNA)– away from the innate sensing system. Therefore, the SARS-CoV-2-mediated modulation of autophagy to generate viroplasms and dsRNA-concealing DMVs may have been a contributing factor for its successful transmission and spread from the natural reservoir to other hosts and finally to humans. However, the underlying mechanisms by which SARS-CoV-2 alters cellular membrane dynamics, and more precisely autophagy, remains to be elucidated. We are currently investigating the role of SARS-CoV-2 in the modulation of autophagy to facilitate virus replication, transmission and spread (Fig. 3). These studies have recently been funded with an NSF RAPID award.
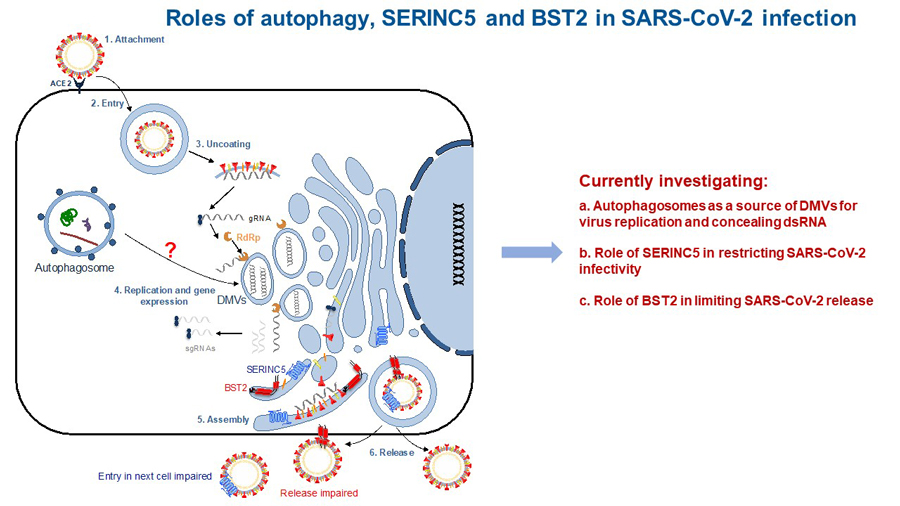
Figure 3
Tetherin and SERINC5 are cellular antiviral defenses with untapped therapeutic potential against SARS-CoV-2 infection. Specifically, these molecules use complementary mechanisms to trap virus particles to the cell surface, preventing (i) the release and spread of nascent virions and (ii) their entry to healthy cells. As a consequence of their prolonged presence on the cell surface, trapped virions become more exposed to the immune surveillance. Despite Tetherin and SERINC5 antiviral properties, the COVID-19 pandemic has evidenced that SARS-CoV-2 is adapted to efficiently bypass human innate barriers. Recent studies have revealed that SARS-CoV-2-infected patients fail at eliciting proper antiviral responses, likely contributing to COVID-19 pathogenesis. Therefore, there is a critical need to uncover the underlying mechanisms by which SARS-CoV-2 counteracts the innate immune system. We are currently investigating how SARS-CoV-2 evades Tetherin and SERINC5 (Fig. 3).