








Normal & Disease-Associated RNA Metabolism
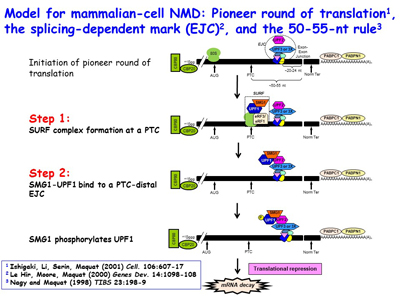
Nonsense-mediated mRNA decay, human disease and disease therapeutics
Research in the Maquat lab utilizes biochemistry, molecular biology, structural biology, genome editing, transcriptomics (e.g. RNA-seq, RIP-Seq, RIP-seq footprinting, NMD-Seq, TRIC-Seq), proteomics and computational biology to study RNA metabolism in human and mouse cells, as well as in the mouse, with a focus on RNA metabolism in human health and disease. As a post-doc, Dr. Maquat was the first to demonstrate that a human disease could be due to pre-mRNA splicing defects, and she also discovered mammalian-cell nonsense-mediated mRNA decay (NMD).
The Maquat lab went on to reveal that NMD, which is a type of mRNA quality control, surveys all newly synthesized mRNAs during what the lab named a "pioneer" round of translation. This round of translation involves mRNA that is associated with the cap-binding protein heterodimer CBP80 and CBP20. It is distinct from the type of translation that supports the bulk of cellular protein synthesis and that involves a different cap-binding protein, eukaryotic translation initiation factor (eIF)4E. The Maquat lab defined the “50-55-nucleotide rule”: generally, if translation terminates more than 50-55 nucleotides upstream of an exon-exon junction that is marked by what the lab called a splicing-dependent “mark”, later called an exon-exon junction complex (EJC), then the mRNA will be subject to NMD. By the time CBP80 and CBP20 have been replaced by eIF4E, the EJC “mark” has been removed so that the mRNA is largely immune to NMD diseases are nonsense-generated.
Selected Awards and Announcements
2024 Albany Prize Awarded to Biochemist Lynne Maquat
Lynne Maquat Receives 2024 Dr. Paul Janssen Award
RNA Biologist Lynne Maquat Awarded 2023 Gruber Genetics Prize
Lynne Maquat Awarded 2021 Wolf Prize in Medicine

Lynne E. Maquat, Ph.D.
Principal Investigator

- Keeping cells fit.; Science (New York, N.Y.); Vol 391(6786), pp. 657-658. 2026 Feb 12.
- TDP-43 dysfunction compromises UPF1-dependent mRNA metabolism in ALS.; Neuron. 2025 Dec 12.
- Gene regulation through exon junction complex modularity.; Nature structural & molecular biology. 2025 Dec 03.
News
Affiliations
- Biochemistry & Biophysics
- Center for Biomedical Informatics
- Center for RNA Biology: From Genome to Therapeutics
- James P. Wilmot Cancer Center
- NIH T32 Training Grant in Cellular, Biochemical & Molecular Sciences
- Biochemistry & Molecular Biology Ph.D. Program
- Biomedical Genetics and Genomics Ph.D. Program
- Biophysics, Structural & Computational Biology Ph.D. Program
August 25, 2025
Fragile X Syndrome: What Happens in the Brain?
May 13, 2025
Congratulations to Victor Gu
April 23, 2025
RNA Discoveries Leading to Cutting-Edge Cures
March 27, 2025
Neuroscience & RNA Biology: How mRNA Decay Shapes Health & Disease
Contact Us
Maquat Lab
MC 3-8507C
601 Elmwood Ave
Rochester, NY 14642
Lynne_Maquat@urmc.rochester.edu
Personal Contact for Dr. Maquat - Lindsay_Vicars@URMC.Rochester.edu
(585) 271-2683