About the Lab
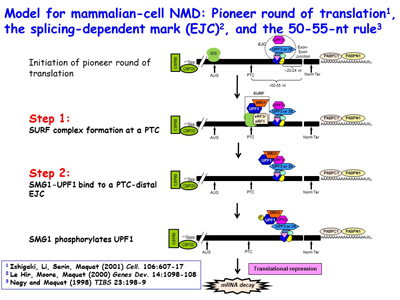
Figure 1. View Enlargement of Figure 1
Nonsense-mediated mRNA decay, human disease and disease therapeutics
View all slides in a Powerpoint file
Research in the Maquat lab utilizes biochemistry, molecular biology, structural biology, genome editing, transcriptomics (e.g. RNA-seq, RIP-Seq, RIP-seq footprinting, NMD-Seq, TRIC-Seq), proteomics and computational biology to study RNA metabolism in human and mouse cells, as well as in the mouse, with a focus on RNA metabolism in human health and disease. As a post-doc, Dr. Maquat was the first to demonstrate that a human disease could be due to pre-mRNA splicing defects, and she also discovered mammalian-cell nonsense-mediated mRNA decay (NMD). The Maquat lab went on to reveal that NMD, which is a type of mRNA quality control, surveys all newly synthesized mRNAs during what the lab named a "pioneer" round of translation. This round of translation involves mRNA that is associated with the cap-binding protein heterodimer CBP80 and CBP20 [Figure 1]. It is distinct from the type of translation that supports the bulk of cellular protein synthesis and that involves a different cap-binding protein, eukaryotic translation initiation factor (eIF)4E. The Maquat lab defined the “50-55-nucleotide rule”: generally, if translation terminates more than 50-55 nucleotides upstream of an exon-exon junction that is marked by what the lab called a splicing-dependent “mark”, later called an exon-exon junction complex (EJC), then the mRNA will be subject to NMD. By the time CBP80 and CBP20 have been replaced by eIF4E, the EJC “mark” has been removed so that the mRNA is largely immune to NMD [Figures 2, 3]. This NMD pathway depends on a 3'-untranslated region (3'UTR) EJC. A much less efficient NMD pathway, initially called failsafe NMD by the Maquat lab, does not involve a 3'UTR EJC and targets both CBP80-bound and eIF4E-bound mRNAs, as first evidenced in mRNA tethering experiments. As does NMD in S. cerevisiae, 3'UTR EJC-independent NMD lasts the life-time of the mRNA.
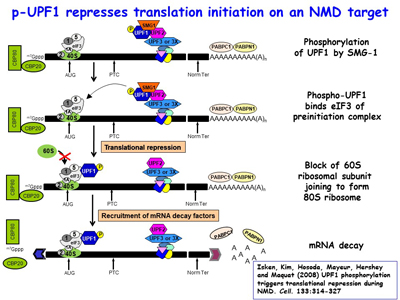
Figure 2. View Enlargement of Figure 2
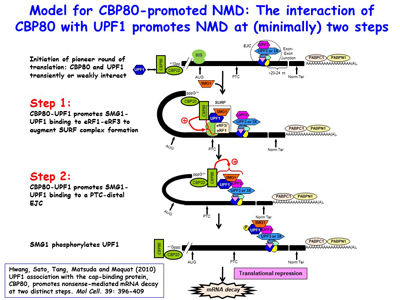
Figure 3. View Enlargement of Figure 3
Subsequent studies defined the mRNA-binding proteins, translation factors and nucleases that trigger NMD. The lab’s most recent work on degradative activities that mediate NMD has revealed ribosome-bound decay intermediates (DIs) with 3'-end nontemplated nucleotides, resolving the long-standing issue of whether decay initiates on polysomes [Figure 4].
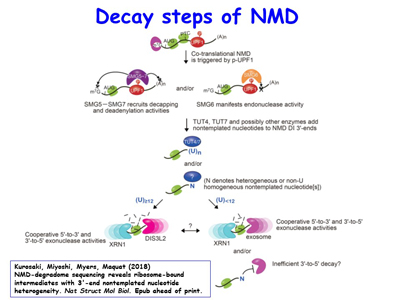
Figure 4. View Enlargement of Figure 4
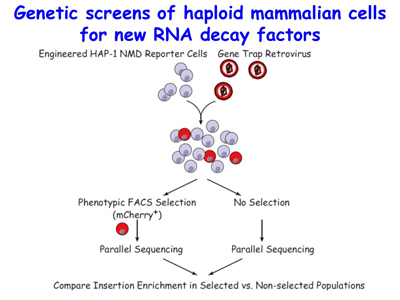
Figure 5. View Enlargement of Figure 5
The lab is now utilizing its mechanistic findings to design and develop therapies to abrogate or promote NMD with the goal of lessening the severity of nonsense-generated diseases. Also ongoing is a genetic screen for new NMD factors using HAP1 haploid human cells [Figure 5].
A remarkable one-third of inherited or acquired diseases are attributable to a nonsense codon that triggers NMD. NMD is much more than a quality-control mechanism. During the course of studies, the Maquat lab has found that NMD is regulated by cells as a means to adapt to changing environments. For example, breast cancer cells given the frontline chemotherapeutic doxorubicin (also sold under the trade name Adriamycin, among others) initiate the caspase-mediated cleavage of the key NMD factor – the ATP-dependent RNA helicase UPF1 – so as to up-regulate the ~10% of normal mRNAs that are natural NMD targets, among which are those encoding pro-apoptotic proteins [Figure 6]. The lab has found that exposing breast-cancer cells to a drug that inhibits NMD, subsequently washing away the drug, and then treating cells with doxorubicin augments the rate and magnitude of cell death. This knowledge may be useful when developing future cancer treatments.
The lab has also found that NMD is hyperactivated in Fragile X Syndrome, which is the most common single-gene cause of inherited intellectual disability. Ongoing work aims to understand the mechanism of inhibition. Tools in hand include induced pluripotent stem cells from control- and FXS patient-fibroblasts that the lab differentiates to cortical neurons [Figure 7].

Figure 6. View Enlargement of Figure 6
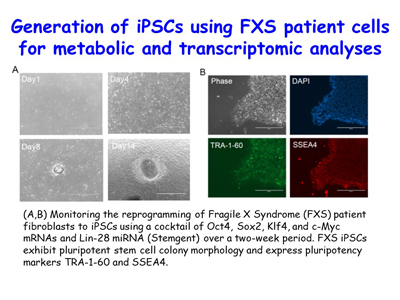
Figure 7. View Enlargement of Figure 7
Staufen-mediated mRNA decay and post-transcriptional effects of Alu elements
Over the past 15 years, the lab’s discovery and subsequent work on the mechanism of Staufen (STAU)-mediated mRNA decay (SMD) has uncovered new roles for cytoplasmic long non-coding RNAs (lncRNAs) and retrotransposon-derived short interspersed elements (SINEs) in post-transcriptional gene regulation. These SINEs include human Alu elements and mouse B1, B2, B4 and ID elements [Figures 8, 9].
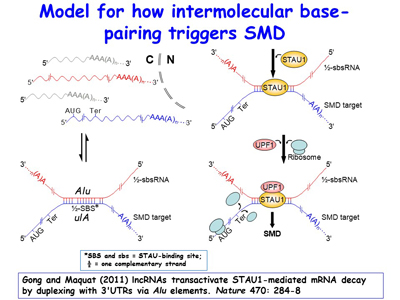
Figure 8. View Enlargement of Figure 8
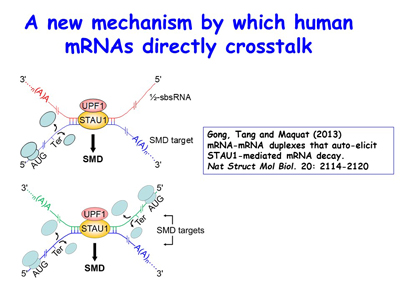
Figure 9. View Enlargement of Figure 9
Ongoing work continues to define new cellular roles for SINEs as sites for nucleating intermolecular base-pairing between different mRNAs, between mRNAs and lncRNAs, and between different lncRNAs. The lab is additionally extending its studies of inverted-repeat Alu elements (IRAlus) and how competitive binding among the many nuclear and cytoplasmic RNA-binding proteins influence nuclear vs cytoplasmic distribution [Figure 10] and the polysome-associated vs. polysome-free (i.e. the translational status) of 3'UTRs that contain one or more Alu elements. Lab members have found that, similar to the benefit of breast-cancer cells inhibiting NMD when given chemotherapeutics so as to promote apoptosis, muscle myoblasts, when deprived of nutrients, inhibit NMD by promoting SMD, which competes with NMD so as promote differentiation into multinucleated myotubes upon nutrient deprivation [Figures 11, 12]. There is an increasing number of scenarios in which the efficiency of NMD is modulated by cells to either maintain homeostasis or differentiate appropriately, depending on the particular stimulus.
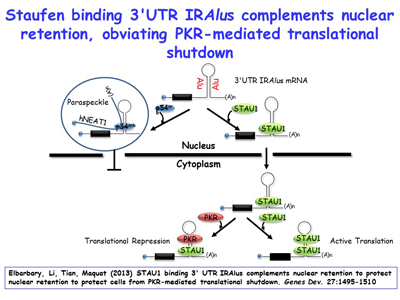
Figure 10. View Enlargement of Figure 10
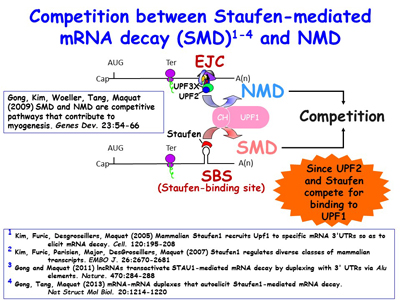
Figure 11. View Enlargement of Figure 11
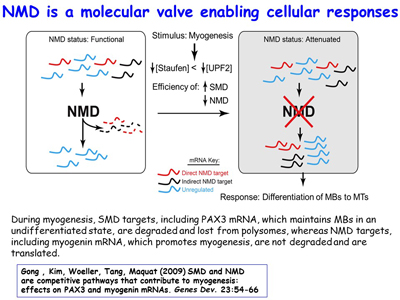
Figure 12. View Enlargement of Figure 12
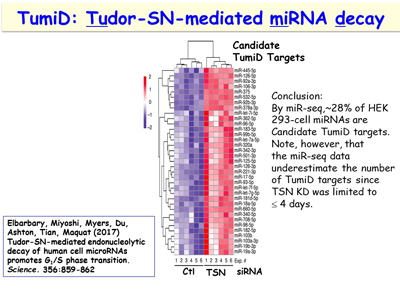
Figure 13. View Enlargement of Figure 13
MicroRNA decay
An additional interest of the lab is following up its discovery of a new microRNA decay pathway that is mediated by the endonuclease Tudor-SN (TSN) [Figure 13]. This pathway, which the lab calls TSN-mediated microRNA decay (TumiD), promotes the G1-to-S phase transition by degrading microRNAs that degrade mRNAs encoding proteins that promote this transition. Like NMD and SMD, TumiD is augmented by the ATP-dependent RNA helicase UPF1 [Figures 14, 15]. Data indicate that UPF1 allows TSN access to scissile CU and AU dinucleotides within a TumiD target by dissociating the miRNA target (while loaded into the RNA-induced silencing complex) from its complementary mRNA. The lab aims to define how TumiD is regulated.
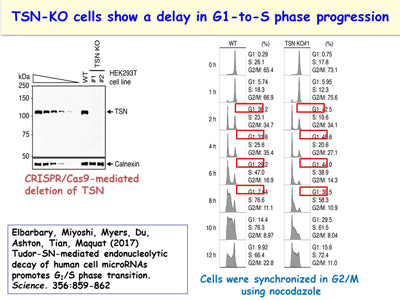
Figure 14. View Enlargement of Figure 14
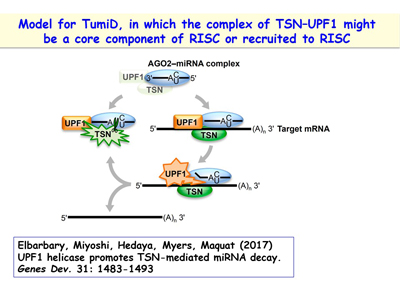
Figure 15. View Enlargement of Figure 15
An unexpected function for CBP80
The lab has uncovered new connections between transcriptional co-activators, the process of RNA synthesis and processing, and the pioneer translation initiation complex. Thus, the lab is currently extending mechanistic links that it has previously established between pre-mRNA splicing in the nucleus and mRNA translation and decay in the cytoplasm to even earlier steps – transcription initiation at gene promoters.
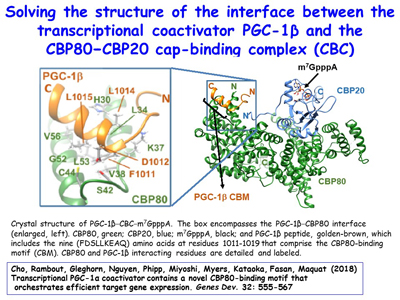
Figure 16. View Enlargement of Figure 16
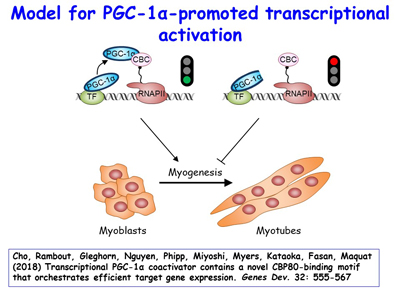
Figure 17. View Enlargement of Figure 17
Although peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1α (PGC-1α) is a well-established transcriptional coactivator for the metabolic adaptation of mammalian cells to diverse physiological stresses, the molecular mechanism by which it functions is incompletely understood. The lab has used in vitro-binding assays, X-ray crystallography, and immunoprecipitations of mouse myoblast-cell lysates to define a previously unknown CBP80-binding motif (CBM) in the C-terminus of PGC-1α [Figure 16]. The CBM, which consists of a nine-amino-acid α helix, is critical for the association of PGC-1α with CBP80 at the 5' cap of target transcripts. Results from RNA sequencing demonstrate that the PGC-1α CBM promotes RNA synthesis from pro-myogenic genes [Figure 17]. These findings reveal a new conduit between DNA-associated and RNA-associated proteins that functions in a cap-binding protein surveillance mechanism, without which efficient differentiation of myoblasts to myotubes fails to occur. The lab has mice generated using CRISPR methodologies to harbor a knock-in of five amino acid changes within the CBM so that PGC-1α no longer binds CBP80 [Figure 18]. Physiological and metabolic studies, coupled with transcriptomics, are underway using skeletal-muscle satellite cells ex vivo.
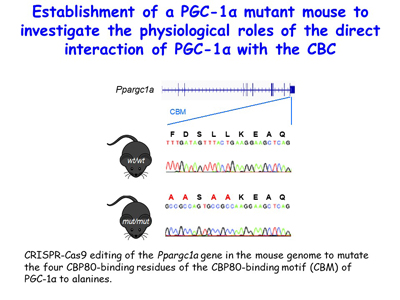
Figure 18. View Enlargement of Figure 18