URMC / Labs / Carpizo Lab / Research Projects
Research Projects
Investigating the mechanism(s) of anti-Netrin-1 therapy in PDAC
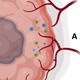 There are no available therapeutics that can reverse EMT and reprogram tumors to a more epithelial phenotype. This project will validate this novel mechanism through a pilot clinical trial in which NP137 (the first in human anti-Netrin-1 therapeutic (Netris Pharma) will be given perioperatively in patients with resectable PDAC.
There are no available therapeutics that can reverse EMT and reprogram tumors to a more epithelial phenotype. This project will validate this novel mechanism through a pilot clinical trial in which NP137 (the first in human anti-Netrin-1 therapeutic (Netris Pharma) will be given perioperatively in patients with resectable PDAC.
Learn more about Investigating the mechanism(s) of anti-Netrin-1 therapy in PDAC
The circadian rythem genes Dec2 and Bmal1 promote cancer dormancy by facilitating immune evasion
 Metastatic recurrence following curative-intent pancreatic cancer (PC) surgery is a major clinical problem as nearly all die of this at both early and latent periods following surgery. Latent recurrences are due to reactivation of dormant tumor cells that disseminate before the primary tumor has been removed. The mechanisms that drive cancer dormancy are poorly understood in part because of the lack of animal models that recapitulate the biology of the resected patient.
Metastatic recurrence following curative-intent pancreatic cancer (PC) surgery is a major clinical problem as nearly all die of this at both early and latent periods following surgery. Latent recurrences are due to reactivation of dormant tumor cells that disseminate before the primary tumor has been removed. The mechanisms that drive cancer dormancy are poorly understood in part because of the lack of animal models that recapitulate the biology of the resected patient.
Learn more about the circadian rhythm genes Dec2 and Bmal1 promote cancer dormancy by facilitating immune evasion
Therapeutic potential and molecular biology of UNC5B in pancreatic ductal adenocarcinoma (PDAC)
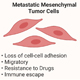 Under some unknown circumstances, primary PDAC undergoes EMT to acquire new phenotypic and cellular features to break away from the primary organ and travel to secondary organs to settle, known as metastasis. Currently, there are no therapeutic or preventive approaches for this phenomenon.
Under some unknown circumstances, primary PDAC undergoes EMT to acquire new phenotypic and cellular features to break away from the primary organ and travel to secondary organs to settle, known as metastasis. Currently, there are no therapeutic or preventive approaches for this phenomenon.
Learn more about Therapeutic Potential and Molecular Biology of UNC5B in Pancreatic ductal adenocarcinoma (PDAC)
Investigating Netrin-1 as a novel therapeutic target in metastatic pancreatic cancer
 Pancreatic cancer is a highly metastatic cancer. The majority of patients with pancreatic cancer are stage IV at the time of diagnosis and the majority of patients (>80%) who have early stage disease go on to suffer from metastatic relapse following curative intent surgery. Novel therapies that target the molecular mechanisms that govern pancreatic cancer metastasis are highly needed.
Pancreatic cancer is a highly metastatic cancer. The majority of patients with pancreatic cancer are stage IV at the time of diagnosis and the majority of patients (>80%) who have early stage disease go on to suffer from metastatic relapse following curative intent surgery. Novel therapies that target the molecular mechanisms that govern pancreatic cancer metastasis are highly needed.
Learn more about Investigating Netrin-1 as a novel therapeutic target in metastatic pancreatic cancer
Investigating the mechanisms of pancreatic cancer dormancy
 The Carpizo lab is at the forefront of research in cancer dormancy having just recently produced the first mouse model of pancreatic cancer dormancy that mimics the biology of the resected patient (bioRxiv 2020.04.13.037374; doi: https://doi.org/10.1101/2020.04.13.037374). This model recapitulates the early and latent recurrent phenotypes seen in human patients following surgery.
The Carpizo lab is at the forefront of research in cancer dormancy having just recently produced the first mouse model of pancreatic cancer dormancy that mimics the biology of the resected patient (bioRxiv 2020.04.13.037374; doi: https://doi.org/10.1101/2020.04.13.037374). This model recapitulates the early and latent recurrent phenotypes seen in human patients following surgery.
Learn more about Investigating the mechanisms of pancreatic cancer dormancy