Frequently Asked Questions
General Questions
RNA/DNA Extractions
QA/QC
High Throughput Sequencing
Epigenomics
Single Cell and Spatial Genomics
Sequencing
Billing
General Questions:
How do I start a study with the GRC?
Prior to starting a study at the GRC, we recommend you contact us through email with a brief description of project goals. We can schedule a meeting to further discuss goals, timelines/deadlines, and budget. Each meeting will involve a member of management, lab personnel, and bioinformatics to ensure all project questions can be addressed. Once you are ready to submit samples, please see our Sample Submission Guidelines page for more information.
How do I get a PPMS ID?
A PPMS ID is required by users submitting samples or for booking user access instruments (qPCR and Covaris). If you do not have a PPMS ID, fill out the RedCap survey to request one. You cannot use the PPMS ID of another user or PI.
How do I submit samples for processing?
Once you have a PPMS ID, please see the Sample Submission Guidelines page for more information.
Can I ship samples to the GRC?
Yes. Please email us to let us know what day you will be shipping your samples. We prefer no shipments be sent on Friday to ensure the package will arrive during normal business hours. If shipping RNA, please package samples securely on dry ice. DNA can be shipped with ice packs. Please send your samples to the following address:
Attention: Elizabeth Pritchett
575 Elmwood Avenue
Wilmot Cancer Center
Room 3-0771
Rochester NY, 14642
Can I bring my samples to the GRC?
Samples can be dropped off in G-7814, near the Biological Supply Center. It is UR-swipe accessible 24/7 and contains a -80C freezer and 4C refrigerator.
Samples are picked up daily between 8am-9am. Please see the Sample Submission Guidelines for more information about sample submission.
What is your typical turn around time?
Turn around time is impacted by our sequencing queues. Our current turn around time is approximately 3 to 6 weeks.
Do you do testing or custom library preparations?
Yes! We often work with researchers to test and optimize custom library preparations. Please email us to schedule a consultation.
Can I use your equipment?
For access to our user access instruments, we require users to book through PPMS. Prior to booking, users should go through a brief training for use of the instruments. Other instruments within the GRC are not available for use by anyone outside of the GRC.
Do you maintain instruments at the GRC?
Yes - we have service contracts with annual preventative maintenance on our instruments to ensure they are performing optimally.
What buffer do you prefer for my samples?
We prefer you elute nucleic acids in nuclease free water. Some buffers (such as TE Buffer) can interfere with downstream processes. If your samples are submitted in a buffer that is not compatible with downstream processes, we will either return the samples or do additional clean ups to replace the buffer (at additional costs).
Do you prefer plates or tubes for sample submission?
For less than 24 samples, we prefer your samples are submitted in 1.5 mL nuclease free tubes. If you have more than 24 samples, we ask that you plate your samples in a 96-well plate in order by column. 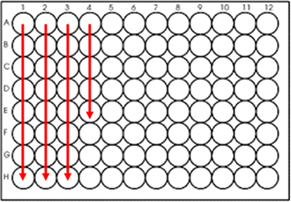
We do not accept samples submitted in 0.5 mL tubes or 8-well strip tubes.
Do you store samples after project completion?
Samples will be stored for 2 weeks following data delivery. We will contact you to schedule sample return. Please see our Policies page.
Does the GRC provide data analysis services?
Yes - the GRC Bioinformatics team provides data analysis services. Please see our bioinformatics service analysis page for more information.
How do I reference the GRC in publications?
All work performed by the University of Rochester Genomics Research Center (GRC - RRID: SCR_012359)) should be acknowledged in scholarly publications, posters, and oral presentations. Proper recognition allows us to measure the impact of our work and supports our initiatives in obtaining sponsored funding. In addition, any GRC personnel who make a substantial intellectual or experimental contribution are deserving of further recognition as co-author on any published material at the discretion of the Director and Principal Investigator.
NCI Designation and Acknowledging the CCSG:
The National Cancer Institute (NCI) named the University of Rochester's Wilmot Cancer Institute as the nation's 73rd designated cancer center. Publicizing the outcomes of NIH-funded projects and communicating the role of NIH support in biomedical research improves public understanding of how we, the biomedical research community as a whole, are working to improve human health. To properly acknowledge the Cancer Center Support Grant (CCSG) in your work which utilized the Shared Resources since March 15th, 2025, please include the following statement in the acknowledgement section of your publication(s):
"The work described in this publication benefited from the support of the Genomics Shared Resource at the Wilmot Cancer Institute, supported in part by the University of Rochester Wilmot Cancer Institute Support Grant #P30CA272302. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health."
Please send citations of any published material to John Ashton and/or Elizabeth Pritchett.
Where can I locate the Institutional Certification document for human genomic data when applying for grants?
The Office of Human Subject Protection and the Research Subjects Review Board (RSRB) have resources to assist. For more information, visit the UR Office for Human Subject Protection Website.
RNA/DNA Extractions
What kits do you offer for extractions?
We offer several kits for the extraction of nucleic acids from many sample types including cells, tissues, serum/plasma, and FFPE, among others. Please see our Extraction page for more information. Ultimately, the GRC will determine which kit is used for your extractions based on sample type, input amount, and desired downstream applications.
What are causes of degraded RNA?
RNA is single stranded and highly susceptible to degradation. Several measures should be considered when extracting RNA:
- Maintain an RNase-free environment. Wear gloves, designate an RNA only workspace, and work quickly when extracting.
- Clean workspace with RNaseZAP - a cleaning solution that will remove RNase
- Use of RNase-free equipment, consumables, and reagents.
- Consider temperature. Extract from fresh tissue or quickly frozen tissue. Maintain RNA at -80ºC and avoid storing at room temperature.
How do you identify if RNA is contaminated with gDNA? How is this resolved?
Increased inter-region area and peaks past the ribosomal peaks often are due to gDNA contamination. To move forward, samples should be treated with DNase to remove this contamination. 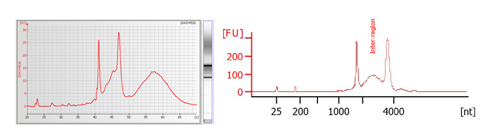
What QC is done after RNA/DNA extraction?
All samples that are extracted by the GRC are assessed for concentration and quality. For RNA, we use Nanodrop and Bioanalyzer. For DNA we use Qubit and Bioanalyzer. To submit samples for extraction and QC, please see our Sample Submission Guidelines page.
What is RLT and RLT Plus Buffer?
RLT Buffer and RLT Plus Buffer are lysing buffers for lysing cells and tissues prior to extraction. RLT and RLT plus are not interchangeable and require different kits. The GRC prefers the use of RLT Plus as this is used with the RNeasy Plus kit that contains a gDNA eliminator column and eliminates the need for DNase treatment.
I would like to submit cells for extraction. How should I submit them to the GRC?
1. The GRC prefers the use of RLT Plus with the RNeasy Plus kits as this includes a gDNA eliminator column. The GRC is able to provide researchers who will submit samples for extraction with RLT Plus (1 mL RLT Plus and 10uL BME).
2. For sorted cells, do not exceed 1/3 volume of RLT Plus (100 uL cells per 350 uL RLT Plus). Vortex well before storing at -80C.
3. Please avoid resuspending in 15mL conicals.
*Visit our extraction page for more information.
QA/QC
How do you determine what QC assays you will use for sample QC?
Many factors influence our decision of QC assay such as sample volume, concentration, and number. The GRC will determine the most appropriate assay for your samples.
What reports do you send with QC?
We send an html QC report that includes all assay results for your samples. If several assays were performed, we will select a concentration source and a trace source (RNA: RIN Source/DNA: Region Source). These choices are listed in the summary section of the report. Following the summary, the individual assays are listed in more detail. The Sample ID is a unique identifier assigned by our online LIMS system. To see an example of the reports that are sent, please visit the QC services page. If you have questions about the report or your results, please contact us.
How do I interpret the different assays?
Please visit the Quality Assessments page for more information on assessment of our QC assays.
What is RIN? RQN?
RIN = RNA Integrity Number from the Bioanalyzer.
RQN = RNA Quality Number from the Fragment Analyzer.
These two values are similar and are used to assess the overall quality of the RNA. Both are on a scale of 1 to 10 with 1 being highly degraded RNA and 10 begin intact high quality RNA. The entire electropherogram is analyzed to determine this value.
Software can often times encounter errors in the trace that prevent it from calculating an accurate RIN. The RIN can be manually adapted to force the software to call an estimated RIN. This RIN is not guaranteed and it is recommended that researchers visually assess the RNA quality before proceeding.
What is the 260/280 ratio? 260/230 ratio?
Both of these ratios are obtained from Nanodrop and are used to determine sample purity.
260 = nucleic acid absorbance
280 = protein absorbance
The 260/280 ratio for pure DNA: ~1.8 and pure RNA: ~2.0.
230 = contaminant absorbance
The 260/230 ratio should be between 2.0 and 2.2.
If these values are lower, contamination should be ruled out. However, these ratios do not necessarily determine success or failure of library preparations.
For more information about contaminants and their impact on Illumina library preparations, click here.
What is QC Only?
QC Only is a service provided for researchers who would like only concentration and quality information for RNA, gDNA, cDNA, NGS libraries. This service requires at least 5 ul of sample and results are sent within 24-48 hours. To submit samples for QC Only, please see our Sample Submission Guidelines page.
High Throughput Sequencing
Do you do testing or custom library preparations?
Yes! We often work with researchers to test and optimize custom library preparations. Please email us to schedule a consultation.
How do I choose the right RNA kit for my sample?
Many factors influence the determination of library preparation kit for samples such as sample concentration, sample quality, and research goals. The GRC will help determine the most appropriate kit for your research goals. Visit our High Throughput Sequencing page for more information about available kits.
What QC is performed on the libraries?
Our typical library QC is Qubit and Fragment Analyzer/TapeStation/Bioanalyzer. All trace assays will provide base pair information for libraries which is used for normalization prior to sequencing. Unless there is a problem with your library preparation, we do not send final library QC results and will proceed with sequencing.
What is Library Normalization?
Prior to sequencing, we normalize all libraries to create an equimolar pool. This ensures we create a well balanced pool and all samples will receive a comparable number of reads. Learn more about library normalization.
What is Library Size Selection?
All of our library preparation methods include a size selection step to capture fragments of certain sizes and to eliminate fragments that are outside the desired size range (such as adapter dimers). Most library size selection steps use Solid Phase Reversible Immobilization (SPRI) technology which captures nucleic acids to magnetic beads such as Ampure XP beads. The range of fragment size that is collected is determined based on the ratio of beads to the sample (see image below). For standard clean ups to remove adapter dimers and small fragments (<200 bp) we use 1X ratio of beads to sample. Some library preparation methods require the need for a double-sided clean up to remove both small and large fragments. Learn more about library size selection in Illumina library preparation methods. 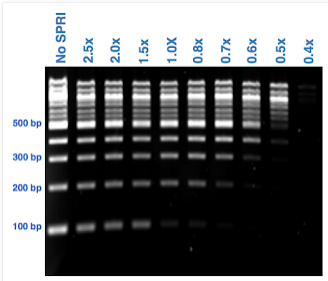
SPRI Ratio Image
Epigenomics
I am interested in ATAC-seq for my favorite cell type. What do I need to know?
We generally require a meeting to walk through the goals of your experiment, determine the biological replicates required, and to schedule a pilot experiment to determine experimental conditions for optimal ATAC library construction. The ATAC library workflow is very sensitive to cell viability and cell number. We require that cells are >85% viable as determined by trypan cell count and/or cell-sorting with a viability stain. Our standard input amount is 50,000 cells per sample, but lower cell numbers are also suitable.
Do you include controls in ATAC experiments?
The GRC will add a gDNA control to each batch of ATAC samples during library prep to assess the activity of the transposase and to help trouble-shoot problematic library preps.
Do you recommend single or paired-end sequencing for ATAC samples?
We recommend paired-end sequencing for ATAC experiments as this allows us to determine library fragment lengths and expands the opportunities for different types of analysis, i.e. nucleosome positioning.
Do you perform Cut and Run or Cut and Tag experiments?
We do not perform the initial steps of the experiment for Cut and RUN/Tag but we do support the library preparation to prepare the samples for sequencing. Please reach out to us for more information.
Do you offer single-cell Multiome?
Yes, we can assist with Multiome experiments that capture the gene expression and ATAC profiles for individual cells. Please contact us for more information.
Single Cell and Spatial Genomics
Which single cell platform is right for my research?
Single cell technology and approaches are rapidly evolving as more researchers being to study single cells. There are many considerations when choosing the most appropriate platform such as procedures for obtaining cells, cell numbers, cell heterogeneity, cost, and overall research goals. Please contact us to schedule a single cell consultation so we can help you decide which approach is most suitable for your research goals.
What is the capture rate of mRNA transcripts per cell for 10X Single Cell?
Depending on reagent chemistry, the capture rate ranges from 6.7 - 8.1% for v1, 14 - 15% for v2 and 30 - 32% for v3. Researchers should keep in mind that this is a stochastic (or random) event and a majority of transcripts are captured when sequencing multiple single cells from the same population.
With the release of the new GEM-X technology, capture rates are improving to nearly 80%. For more information, contact us.
What impact does cell viability have on 10X single cell experiments?
Cell viability is a critical component of 10X Single Cell experimental success. Non-viable cells decrease overall application performance and lead to decreased library complexity and inaccurate cell counts. Dead cell removal is recommended for samples with less than 70% viability.
Is there evidence of technical variation between captures and/or capture wells?
10X conducted a study to assess the potential for technical variation in 10X experiments. In this study, 10X investigated the potential impact of the following:
1) Captures on 2 different chips
2) Captures in 2 different wells of the same chip
3) Sequencing of captures on 2 separate flow cells
This study indicates highly correlated expression profiles and near-identical cell clustering between each of the tested scenarios.
Do you have requirements for submission of sorted cells for single cell such as buffers and tube types?
We ask that you sort your cells into 1.5mL tubes in the media/buffer that keeps your cells the most happy. There are some buffers that are problematic:
- FBS > 10%
- BSA > 2%
- EDTA > 0.1mM
- Tween 20
- Mg. Ca > 3mM
Do you recommend flow sorting my cells prior to capture?
Flow sorting can be a viable option to isolate your cells of interest and remove dead cells and debris. If you plan to sort your cells, please click here to see a 10X bulletin discussing best practices for a capture.
Do you offer feature barcoding approaches such as Hashtag cell labeling and/or CITEseq?
Yes! We help coordinate feature barcoding projects for single-cell experiments. We stock many TotalSeq antibodies from BioLegend to help with hashtag cell labeling experiments. Please reach out to us to start the conversation!
Do you offer the latest reagent kits from 10X Genomics such as Fixed RNA profiling and GEM-X?
Yes. We continue to offer our research community the latest in single cell technologies. This includes Fixed RNA Profiling and the newest GEM-X reagents.
Do you offer Spatial Genomics?
Yes - the GRC has used this technology for many tissues types. Please contact us for more information about this platform and how you can start a Spatial Transcriptomics study.
How do I assess if my Fresh Frozen tissue blocks are suitable for Spatial Genomics using the Visium platform?
10X currently has two Visium options: Fresh Frozen and FFPE. Before beginning optimization or gene expression work with the Fresh Frozen Visium platform, it is important to assess the RNA quality of the tissue blocks you intend to use. To do this, please section 10 sections at 10um for RNA extraction and RNA QC to determine the RNA Integrity Number, or RIN, of your tissue sections. The sections can be placed in a pre-chilled 1.5mL tube and stored at -80ºC. 10X recommends using tissue blocks with RIN values greater than or equal to 7.
Is the Visum platform compatible with FFPE tissue blocks?
Yes! 10X genomics has officially released the Visium for FFPE workflow and the GRC is currently testing this internally. It is currently compatible with human and mouse FFPE tissues.
What type of tissue block is compatible with Visium for FFPE?
Human or mouse tissues should be fixed in a crosslinking formaldehyde solution, such as 10% neutral buffered formalin (NBF) or 4% paraformaldehyde (PFA), and then embedded in paraffin wax.
Are there any RNA quality checks on FFPE blocks prior to starting a Visium study?
Yes. 10X recommends extracting RNA to determine the DV200%. This measures the percentage of RNA fragments that are greater than 200 nt. 10X recommends proceeding with FFPE blocks with a DV200% of 50 or greater. To extract RNA, please submit the following:
- For small tissues (less than or equal to 6.5 x 6.5 mm), section 4 (10um) sections in microcentrifuge tube
- For larger tissues (greater than 6.5 x 6.5 mm), section 1-2 (10um) sections in microcentrifuge tube
Do you have the NanoString GeoMx platform?
Yes! The GRC has the NanoString GeoMx platform and is ready to offer this service to the research community. Please contact us to schedule a meeting to discuss this approach.
Do you have the 10X Xenium in situ platform?
Yes! We recently acquired the Xenium instrument to help provide a targeted spatial solution for the community. Please contact us to schedule a meeting to discuss this approach.
Sequencing
How does Illumina sequencing work?
Illumina technology uses Sequencing by Synthesis (SBS). Learn more about Illumina sequencing technology by watching Illumina's brief sequencing overview video.
Which sequencer is right for my research?
The Genomics Research Center will determine the most appropriate sequencer based on sequencing queue and necessary read depth for the project.
With the acquisition of the new NovaSeq X PLUS, we offer flow cell sharing for researchers with a paired-150 bp configuration.
What kit sizes are available on the NovaSeq 6000?
With the acquisition of the NovaSeq X PLUS, we have transitioned our NovaSeq 6000 runs to the new instrument. We are able to run specific flow cells, if requested, but recommend use of the new instrument.
What kit sizes are available on the NextSeq 2000?
P1 (Spec: 100 Million Reads):
- 100 cycles
- 300 cycles
- 600 cycles
P2 (Spec: 400 Million Reads):
- 100 cycles
P3 (Spec: 1.2 Billion Reads):
- 100 cycles
*Please note: There are additional flow cell options, please inquire if interested. The NextSeq flow cells support both paired-end and single-end runs.
What kit sizes are available on the new NovaSeq X PLUS?
NovaSeq 1.5B (Spec: 1.5 Billion Reads):
- 300 cycles (2 x 150)
- 600 cycles (2 x 300)
NovaSeq 5B (Spec: 5 Billion Reads):
- 100, 200, 300 cycles
NovaSeq 10B (Spec: 10 Billion Reads)
- 300 cycles (2 x 150)
- 200 cycles (2 x 100)
- 100 cycles (2 x 50)
NovaSeq 25B (Spec: 25 Billion Reads):
- 300 cycles (2 x 150)
*Please note: There are other flow cell options available. Please inquire if interested. The NovaSeq flow cells support both paired-end and single-end runs. For example, a 100 cycle kit will contain enough reagents to run single-end 100 OR paired-end 50.
What is 2-Channel vs 4-Channel Chemistry on Illumina sequencing instruments?
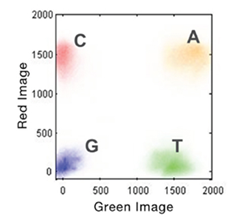
4-Channel chemistry is used for the MiSeq and HiSeq instruments while NextSeq and NovaSeq use 2-Channel chemistry. For the 4-Channel instruments, each nucleotide is represented by a separate dye. Each sequencing cycle requires 4 images to capture this information (Figure 1). With 2-Channel instruments, only 2 dyes are used: red and green. Figure 2 shows the imaging output for the 4 bases:
A= red + green
C = red
T = green
G = dark
Implementation of the 2-Channel chemistry allows for only 2 images per sequencing cycle which leads to faster sequencing and data processing. 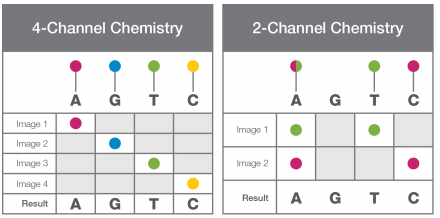
NovaSeq X Plus 2-Channel Chemistry:
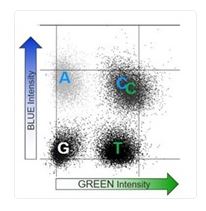
Figure 1 Figure 2
Learn more about Illumina Image Chemistry.
2-Channel chemistry is more susceptible to issues with base diversity within a library. Nucleotide balance is important for color matrix correction and intensity normalization. If a particular library has a stretch of G nucleotides, the imaging will be dark and may cause the run to fail. Several measures are taken if a library is suspected to have low nucleotide diversity. Please scroll down to our FAQ about library diversity and its impacts on sequencing.
The NovaSeq X PLUS instrument has 2-channel chemistry but is a different configuration than the NovaSeq 6000. What implication does this have on sequencing?
The NovaSeq X PLUS contains blue and green channels. It is recommended to utilize index sequences that contain both channels for every cycle, if possible. Illumina recommends avoiding index combinations which only have signal in the blue channel from A or A+G in any given cycle, or no signal (only G) present in any given cycle. For more information on the channel updates for the NovaSeq X PLUS, click here.
The NextSeq 2000 and NovaSeq X PLUS utilize XLEAP chemistry. What is this and how has it improved sequencing?
XLEAP chemistry involves several reagent improvements that allow for faster cycling, higher fidelity, and an overall increase in stability that leads to increased base calling accuracy. These important improvements build upon the Illumina SBS chemistry but allow for faster turnaround and lower costs for our users. We are proud to be the only center in the region to offer services on this groundbreaking instrument. For more information on the improvements, please watch this youtube video from Illumina. Contact us for more information!
What is the difference between single and paired end sequencing?
Single end sequences all fragments from one end while paired end sequences from both ends of a fragment. Paired end reads allow for increased mapping accuracy and isoform detection. Standard differential gene expression studies often require only single end reads. Paired end sequencing is beneficial when identifying genomic rearrangements, gene fusions, insertions, deletions, and repetitive sequences (among others).
Learn more about Illumina paired-end sequencing.
I was quoted for PER50 for each library. Can you explain what this means?
1 cluster is the clonal amplification of a cDNA molecule forming a spot on the flow cell. 1 cluster can generate a read 1 (single-end sequencing) or a read 1 and read 2 (paired-end sequencing). For this particular example, 50 million clusters = 50 million read 1 + 50 million read 2.
Can you sequence libraries I created in my lab?
Yes. Please visit our Sample Submission Guidelines page for submission of User Prepared Libraries. Please indicate the index sequences used in your preparation and if any custom sequencing primers are needed. All libraries submitted for sequencing will have QC to ensure the samples are ready for sequencing. While we do offer sequencing of user prepared libraries, we cannot guarantee sequencing success on library preparations performed outside of the GRC and you will be responsible for the cost regardless of the outcome. Please access the Illumina Adapter Sequences document to design libraries that are compatible with Illumina sequencers.
What are primer dimers? Adapter dimers? Why are they an issue with sequencing?
Primer dimers are when the primers ligate to one another and cause a peak < 100 bp. Adapter dimers are when the P5 and P7 adapters ligate to one another and cause a peak at ~125bp. Primer dimers do not form clusters and will not be sequenced but they can bind to the flow cell surface and will lead to decreased yield. Adapter dimers are able to form clusters and sequence very efficiently and may lead to index hopping. If these peaks are present, an ampure clean up should be done to remove these prior to sequencing. Learn more about adapter dimers and the impact on sequencing. 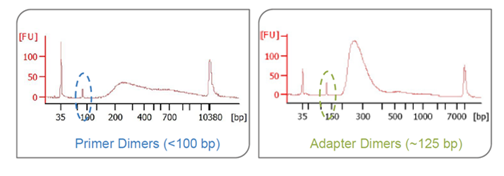
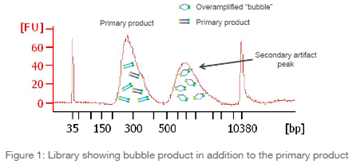
What is a bubble product?
A bubble product is a partially open bubble-like conformation that is comprised of double stranded adapter sequences flanking single stranded noncomplementary inserts. These bubble products can be a result of too much input or too many PCR cycles. Libraries that contain a bubble product can be successfully sequenced. Bioanalyzer analysis is important for identifying libraries that contain bubble products. Learn more about bubble products.
What is index hopping?
Index hopping is the misassignment of sequencing reads to the wrong index. This occurs more often on the patterned flow cells, like those used on the NextSeq and NovaSeq. Index hopping can be corrected by eliminating free adapters and using unique dual indexes.
Learn more about index hopping in Illumina's whitepage.
What are UDI Indexes?
Illumina offers Unique Dual Indexes (UDI). This provides 96 unique dual indexes for sequencing which allows for more multiplexing (number of samples in a pool) and helps correct index hopping. Any combinations that are unexpected will be removed informatically. The GRC has converted to using these indexes for our library preparations whenever possible. For shared runs on our NovaSeq X, we require users utilize the UDI strategy.
Learn more about Illumina's unique dual indexes.
What is PhiX?
PhiX is a small, well characterized bacteriophage genome. It consists of a well balanced base composition with ~45% GC and ~55% AT. Because of this, it is used as an in-run control for run quality monitoring.
Learn more about PhiX.
What is library diversity? What impact does it have on sequencing?
Library diversity is the composition of nucleotides present within the library. For libraries with a well-balanced nucleotide composition, we spike-in 1% PhiX as an in-run control. If libraries are expected to have low diversity, we will increase the percent of PhiX to help with diversity and sequencing success.
Learn more about nucleotide diversity and its impacts on sequencing.
My 16S libraries have poor sequencing quality, how can I improve this?
16S libraries can have low library diversity which can negatively impact sequencing quality. To help with this, you can incorporate heterogeneity spacers (aka Phased Primers) to increase the library diversity and sequencing quality. Click here for a helpful resource on designing these spacers.
Can I pool different library types for sequencing?
It is not recommended to pool different library types for sequencing. It is difficult to determine loading concentration for pools with varying library sizes as these libraries have different clustering efficiencies. For example, smaller libraries will cluster much more efficiently than larger libraries and will be overrepresented within the run. It is recommended to pool libraries prepared with the same library preparation method. However, if libraries are generated with similar strategies and share similar sizing, we are able to pool them together as we have optimized performance on the NovaSeq X PLUS. We require users utilize the Unique Dual Indexing strategy.
Learn more about pooling different library types.
If I do not need a full sequencing run, can I add my samples to another investigators sequencing run?
With the addition of the NovaSeq X PLUS, lane sharing is now a possibility to help researchers reduce sequencing cost. The Genomics Research Center has optimized shared sequencing runs by combining similar libraries that contain compatible Unique Dual Indexes. We ensure we are combining libraries that will perform well with one another and have sufficient index diversity. For user prepared libraries, we will do our best to determine the most appropriate loading and pooling ratio but we cannot guarantee that the desired read depth will be met.
Billing
Can I pre-bill for services?
We are unable to pre-bill anyone for services. Pre-billing for reagents is possible at the discretion of Elizabeth Pritchett or John Ashton. Please contact us for more information.
Do you offer additional discounts?
The Genomics Research Center is subsidized by the University of Rochester and additional discounts are not available to anyone other than Wilmot Cancer Center members.
When will I receive my bill?
We generate an invoice on the first day of the month following the completion of the project.
Can I change the account number to be charged after I submit my samples?
Yes, up until 5 days after the DRAFT invoice is sent out.
Who will receive the invoice?
The PI and any group administrators designated in PPMS.
How do I change my PI and/or account?
Use the PPMS link to make changes to your PPMS information.
Why does the invoice contain the wrong account number?
The invoice is generated using the account number listed on the submission sheet. If no account number is submitted, the PPMS default account will be used. The account number can also be changed up until it is finalized by emailing urgenomics@urmc.rochester.edu.
Can I use a credit card to pay an invoice?
Yes! We can now accept credit card payments for invoices. Once you receive your invoice, you will be able to select the credit card link to complete the purchase.